


Цитогенетическая и молекулярно-цитогенетическая характеристика микроделяционных синдромов Прадера-Вилли, Ангельмана и Ди Джорджи
Одной из наиболее актуальных проблем современной медицинской генетики является определение этиологии и патогенеза наследственных заболеваний. Цитогенетические и молекулярные исследования имеют высокую диагностическую информативность и ценность при решении данной проблемы, так как хромосомные аномалии встречаются с частотой от 4 до 34% при различных наследственных синдромах. В последние годы появились новые методы цитогенетических и молекулярно-цитогенетических исследований, значительно расширяющие диагностические возможности при заболеваниях, сопровождающихся различными хромосомными нарушениями. Данный обзор посвящен вопросам выбора необходимых цитогенетических или молекулярно-генетических анализов при различных формах генетических синдромов.
В работе рассматриваются генетические заболевания, вызванные микроделециями хромосом. На примере наследственных заболеваний (синдромах Прадера-Вилли, Ангельмана, Ди Джорджи) будет изучено явление геномного импринтинга, влияние которого ученые изучают и сейчас, так как до конца механизм геномного импринтинга пока не известен. Будут рассмотрены возможные варианты медицинской помощи при данных заболеваниях и риски дальнейших проявлений этих заболеваний в семьях, где встречаются такие патологии. Изучение этой проблемы на сегодня является актуальной.
Целью работы является: изучение цитогенетических и клинических проявлений микроделяционных синдромов Прадера-Вилли, Ангельмана и Ди Джорджи.
ГЛАВА 1. СОВРЕМЕННЫЕ МЕТОДЫ И ПРОБЛЕМЫ ДИАГНОСТИКИ НАСЛЕДСТВЕННОЙ ПАТОЛОГИИ
1.1 Современные представления о наследственных заболеваниях
Одной из наиболее актуальных проблем современного здравоохранения является организация медико-генетической помощи семьям, где встречаются случаи рождения детей с наследственными заболеваниями. Актуальность этой проблемы определяется широким распространением данной патологии, трудностью дифференциальной диагностики большого количества наследственных болезней, проявляющихся множественными аномалиями развития с высоким риском повторения заболевания в семье.
По данным, опубликованным в начале 90-х годов прошлого века, в мире насчитывалось около 7,5 млн. человек с наследственными патологиями, одним из клинических проявлений которых является умственная отсталость. Цитогенетические методы исследования играют огромную роль в выяснении этиологии генетических заболеваний. Согласно последним сообщениям, хромосомные аномалии среди пациентов с задержкой развития и умственной отсталостью встречаются в среднем с частотой от 4 до 34% (1, 3).
В настоящий момент появились новые методы цитогенетического и молекулярно-цитогенетического исследований, значительно расширяющие спектр современных диагностических возможностей. К этим методам можно отнести FISH-метод, основанный на гибридизации флюоресцентно меченных ДНК-зондов на разные участки генома; сравнительную геномную гибридизацию; метод спектрального кариотипирования; праймерное исследование in situ и др. В связи с разнообразием современных цитогенетических методов в литературе широко обсуждается вопрос о выборе необходимого цитогенетического или молекулярно-генетического анализа для установления диагноза при задержках психического развития и умственной отсталости (4).
В соответствии с рекомендациями согласительной конференции американской коллегии медицинской генетики (The Consensus Conference of the American College of Medical Genetics, 1997), любому пациенту с такой патологией на первом этапе необходимо провести стандартный цитогенетический анализ c разрешением в 500 сегментов. В случае подозрения на синдром, обусловленный микроструктурными аномалиями, необходимо проведение более чувствительных молекулярно-цитогенетических исследований. Первые микроструктурные нарушения хромосом, ассоциированные с определенным синдромом, были обнаружены в 1980 г. Е. Buhler и соавторами. Они сообщили о терминальной делеции сегмента 8q24 у девочки с клиническими признаками синдрома Лангера-Гидеона. К настоящему времени насчитывается около двух десятков микроделеционных синдромов, обусловленных терминальными и интерстициальными делециями разных хромосом (Рубинштейна-Тейби, Миллера-Дикера, Вильямса, Лангера-Гидеона, Прадера-Вилли, Ангельмана, Аладжилла, Ди Джорджи, Рассела-Сильвера и др.) (5, 9-12).
Микроделеции и микродупликации обычно затрагивают ряд близко сцепленных генов, доза которых в результате такой перестройки существенно меняется. В 1986 г. R. Schmickel предложил обозначать болезни, обусловленные изменением дозы близко расположенных генов в результате микроделеций или микродупликаций, термином «синдром смежных генов» (contiguous gene syndrome). Очень важно, что идентифицируются данные синдромы еще до цитологической локализации, и это в свою очередь является главным критерием их обозначения как «синдрома смежных генов». В литературе описаны случаи таких заболеваний, обусловленные не только делециями и дупликациями хромосомных сегментов, но и без них. Указанные синдромы являются преимущественно спорадическими. Отмечено, что размер вовлеченного в хромосомную перестройку района связан с тяжестью заболевания (7).
Многочисленные сообщения последних лет свидетельствуют о существенной роли субтеломерных перестроек в генезе недифференцированной умственной отсталости. Показано, что субтеломерные регионы хромосом насыщены генами, и мутации в них могут приводить к генетическим нарушениям. В настоящий момент анализ субтеломерных перестроек, проводимый различными методами, был выполнен во многих выборках пациентов с наследственными заболеваниями, у которых было выражено отставание в развитии. Фактически субмикроскопические субтеломерные хромосомные аномалии были обнаружены у 6,5-7,4% детей с умеренной и тяжелой умственной отсталостью (16, 17) и у 10,3% детей с легкой умственной отсталостью. Благодаря этим исследованиям были описаны следующие формы патологии:
- субмикроскопическая терминальная делеция 8pter, связанная с транслокацией t(8;20), приводящая к психическим и поведенческим проблемам;
- терминальная делеция хромосомы 5р у пациента с фенотипическими проявлениями синдрома Lujan-Fryns;
- тандемная транслокация 22/15 с делецией района 22q13.3 и сохранением района ядрышкового организатора хромосомы у пациента с задержкой психомоторного и речевого развития и гипотонией, без каких-либо дисморфологических особенностей;
- делеция района 22q13 у пациентов с задержкой психомоторного развития и речи, гипотонией и незначительными малыми аномалиями;
- делеция 16р, возникшая de novo, сочетающаяся с гипотонией и неспецифическими аномалиями; - субтеломерная делеция вследствие семейной сбалансированной транслокации t(3;16) (q29; р13.3), сегрегирующей в двух поколениях;
- делеция района 1р36.3 (выполнен комплексный геномный анализ карт сцепления) (2, 4). Данная хромосомная аномалия может быть связана с гипотонией, аномалиями роста, характерным лицевым фенотипом (выпуклый лоб, глубоко посаженные глаза, плоская переносица, гипоплазия средней трети лица и выступающий подбородок), кардиомиопатией, расширением желудочков мозга, гипоплазией мозолистого тела, лейкодистрофией, психическими расстройствами (4).
Данные случаи еще раз указывают на важность выявления субтеломерных микроперестроек при спорадических и семейных формах генетических заболеваний. В связи с техническими сложностями и высокой стоимостью исследования субтеломерных участков предварительно необходим тщательный клинический отбор обследуемых пациентов. С этой целью B. de Vries и соавторы обследовали 29 пациентов с уже известной субтеломерной аномалией и оценили их клинические данные, семейный анамнез, анамнез родов, лицевые дисморфологические признаки и врожденные пороки (4). Контрольную группу составили 110 детей с умственной отсталостью неясной этиологии, но без субмикроскопических субтеломерных перестроек. На основании этих исследований были разработаны показания для направления пациентов на исследование субмикроскопических субтеломерных перестроек. Такими показаниями являются семейный характер умственной отсталости; пренатальная гипотрофия; постнатальная задержка или опережение физического развития; наличие не менее двух малых аномалий и одной или более «нелицевой» малой аномалии и/или врожденного порока развития. В настоящий момент нет единого мнения относительно правильности и полноты этих рекомендаций. Для повышения эффективности диагностики субтеломерных нарушений необходимо выявление детальных клинических характеристик и применение методов исследования генома (4). В последнее время появились новые методы, позволяющие измерить число копий локуса с помощью гибридизации со специфическим набором проб на концевых участках хромосом. С. Sismani и соавторы сообщили о так называемом MAPH-методе, использованном ими для скрининга с целью выявления субтеломерных перестроек. Этот метод является более быстрым и экономически выгодным, что позволяет рекомендовать его для скрининга субтеломерных перестроек. Отдельную проблему в медико-генетическом консультировании представляют пациенты с подозрением на Х-сцепленные формы генетических патологий, которые встречаются в популяции со средней частотой 0,15%. В последнее время на хромосоме Х было идентифицировано несколько генов, ответственных за неспецифическую умственную отсталость: мутации в гене TM4SF2 interleukin-1 семейства рецепторов IL1RAPL1 и в гене VCX-A; мутации гомеобоксного гена ARX и гена L1CAM при Х-сцепленной гидроцефалии. Недавно описан синдром МЕНМО включающий в себя умственную отсталость, эпилептические приступы, гипогенитализм, микроцефалию и ожирение. Ген, определяющий это состояние, локализован в районе Хр21.1-р22.13. Наиболее распространенной формой Х-сцепленной умственной отсталости является синдром Мартина-Белла, или синдром фрагильности Х-хромосомы (FRA-X). Известно, что частота синдрома Мартина-Белла в общей популяции мальчиков составляет 0,05-0,025%, а среди умственно отсталых пациентов мужского пола его частота значительно выше и колеблется, по разным данным, от 1,6 до 22%. Новые возможности для диагностики синдрома Мартина-Белла появились с развитием молекулярно-генетических методов исследования. Оказалось, что причиной возникновения синдрома является экспансия тринуклеотидного повтора CGG в генах FMR1 и FMR2. Много сообщений подтверждают общий клинический опыт о необходимости хромосомного анализа у пациентов с неспецифической, несиндромальной умственной отсталостью для выявления анеуплоидии по половым хромосомам и FRA-X. В исследовании А. Battaglia и соавт. у 10,2% детей с задержками развития была выявлена анеуплоидия, а у 5,1% была диагностирована FRA-X. М. Khalifa и соавт. обследовали 1205 пациентов с умственной отсталостью и выявили среди них 8 мальчиков с синдромом Клайнфельтера и 3 мальчиков с синдромом FRA-Х. В 1999 г. впервые была описана, а затем подтверждена другими исследованиями мутация в гене метил-CpG связывающего белка 2 (MECP2), вызывающая синдром Ретта (4).
Таким образом, для выявления генетических патологий, в настоящее время, стали широко использовать не только цитогенетические, но и молекулярно-цитогенетические методы исследований. Это позволяет более полно изучить проблемы наследственных заболеваний.
1.2 Геномный импринтинг
В середине XIX в. Грегор Мендель в своих опытах по скрещиванию гороха сделал наблюдение, которое впоследствии стало настоящей аксиомой для генетиков. Он обнаружил, что, если скрестить гомозиготное растение, имеющее гладкие семена, и гомозиготное растение с морщинистыми семенами, в потомстве все растения будут одинаковыми и дадут только гладкие семена. Этот результат не зависел от того, у какого из растении, взятых для скрещивания, - мужского или женского - семена были гладкими. Так Мендель открыл принцип эквивалентности реципрокных скрещиваний: у потомства ген действует одинаково независимо от того, от кого из родителей он унаследован.
Трудно переоценить значение этого наблюдения Менделя в истории и практике генетики. Было установлено, что такой закономерности подчиняется большое число наследственных признаков - не только у гороха, но и у многих других организмов (3).
Исключения из правила идентичности гибридов при реципрокных скрещиваниях (т. е. скрещиваниях между двумя формами, когда каждая из них в одном случае берется в качестве матери, а в другом - в качестве отца) в действительности известны давно, однако, как правило, их можно было отнести к одному из двух классов. Первый составляют признаки, которые определяются генами, расположенными в половых хромосомах: у самок млекопитающих в ядрах клеток имеется по две Х-хромосомы, у самцов - по одной Х- и одной У-хромосоме. Например, цветовая слепота и гемофилия связаны с генами X-хромосомы. Наследование этих сцепленных с полом признаков подчиняется вполне определенным правилам, согласно которым гибриды в реципрокных скрещиваниях не обязательно идентичны. Рассмотрим пример: у отца-дальтоника и нормальной по этому признаку матери ни один из сыновей не будет дальтоником. Если мать страдает дальтонизмом, а отец нет, то все сыновья окажутся дальтониками. В обоих случаях дочери будут нести ген, обусловливающий дальтонизм, но иметь нормальное зрение. Наследование и проявление признаков, сцепленных с полом, зависят от пола потомка, но не связаны непосредственно с полом того родителя, от которого унаследован признак.
Второй класс неэквивалентных реципрокных скрещиваний включает признаки, определяемые внеядерными генами. Некоторые клеточные органеллы - митохондрии в клетках животных, митохондрии и хлоропласты в клетках растений - обладают своей собственной генетической информацией. Эти органеллы передаются из поколения в поколение с цитоплазмой яйцеклеток и поэтому наследуются исключительно по материнской линии. Таков характер наследования цвета листьев у некоторых растений, а также заболевания человека, известного под названием митохондриальной энцефаломиопатии. При митохондриальном наследовании зигота, образующаяся в результате слияния половых клеток, получает митохондрии и содержащуюся в них мтДНК только через яйцеклетку (3).
Недавно генетики и эмбриологи описали третье исключение — это геномный импринтинг, когда оба родителя передают потомкам совершенно идентичные гены, но эти гены несут специфический отпечаток пола родителей, т.е. отцовские и материнские гены активированы или супрессированы во время гаметогенеза по-разному. Таким образом, в некоторых случаях важно, от кого из родителей унаследован ген (1). Суть геномного импринтинга заключается в том, что гены, передаваемые потомству, несут специфический «отпечаток» пола родителя, т.е. отцовские и материнские гены маркированы по-разному; причем эти «отпечатки» временные и могут быть «стерты». Вследствие геномного импринтинга потомки, получившие маркированные гены от матери, отличаются от тех, которые унаследовали такие гены от отца. Другими словами, в некоторых случаях важно, от которого из родителей унаследован ген (1, 3).
Многие исследователи пытались установить молекулярную природу геномного импринтинга, обеспечивающие его механизмы, а также число и функции маркируемых генов. Благодаря этому сделано несколько замечательных открытий, которые расширяют понимание ряда раковых и наследственных заболеваний, а также некоторых других патологий. Изучение геномного импринтинга, возможно, откроет что-то новое и в наследовании признаков, которые вполне удовлетворительно объясняются в рамках классической менделевской генетики (3).
Термин «импринтинг» (imprint — отпечаток) впервые предложил в 1960 г. Х. Кроуз из Колумбийского университета США для описания селективной элиминации отцовских хромосом у насекомых. Геномный импринтинг называют эпигенетическим явлением, подчеркивая этим, что наследуются изменения генной активности, обусловленные родительским происхождением хромосом или их фрагментов, а не структурные перестройки генетического материала (мутации). Таким образом, в некоторых участках генома, подверженных геномному импринтингу, экспрессируется только один отцовский или материнский аллель, т.е. наблюдается моноаллельная экспрессия импринтированных генов (генов, которые дифференциально экспрессируются в зависимости от отцовского или материнского происхождения) в отличие от обычной диаллельной. Причем, если импринтирован материнский ген, то экспрессируется отцовский аллель и наоборот. Наличие такого способа регуляции работы генов свидетельствует о неэквивалентном вкладе родителей в функционирование генома потомков, а фенотипические признаки, контролируемые импринтированными локусами, могут появляться в результате не только мутаций генов, но и нарушения эпигенетической программы регуляции генной экспрессии (1).
Геномный импринтинг занимает особое место среди специфических механизмов регуляции активности генов на ранних стадиях развития, приводя к различиям в экспрессии гомологичных материнских и отцовских аллелей. Первоначальный «отпечаток», созданный в половых клетках, служит основанием для дальнейших модификаций в результате взаимодействий между родительскими геномами и цитоплазматическими факторами яйцеклетки во время формирования пронуклеуса (автономное существование яйцеклетки и сперматозоида в зиготе). Последующие эпигенетические модификации могут привести к тому, что изменения в экспрессии генов будут стабильно передаваться в процессе развития клеточных поколений. Геномный импринтинг, например, может изменять дозу генов, контролирующих рост эмбриона, клеточную пролиферацию и дифференцировку (1).
Изучение геномного импринтинга у млекопитающих началось в начале 80-х годов XX в. после опытов на мышах, проведенных Дж. Мак Гратом, Д. Солтером и М. Сурани (рис. 1). Авторы разработали тонкий микрохирургический метод переноса клеточных ядер мышиных эмбрионов в стадии пронуклеусов и показали, что наследование хромосомных наборов только от одного из родителей приводит к нарушению процесса развития. Оказалось, что отцовский генетический вклад важен для развития плаценты, а материнский вклад необходим для развития тела эмбриона. Далее было показано, что наследование части индивидуальных хромосом или целой хромосомы только от одного из родителей может также приводить к аномальному фенотипу (3).
Таким образом, геномный импринтинг состоит в том, что хромосомы половых клеток (сперматозоидов или яйцеклеток) индивида приобретают «отпечаток» его пола (рис. 2). Потомство получает один набор хромосом с отцовской маркировкой некоторых генов, а другой - с материнской. При образовании у потомка половых клеток прежний «отпечаток» стирается и эти гены маркируются в соответствии с полом данной особи.
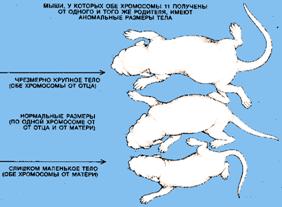
Рис. 1 Геномный импринтинг у мышей
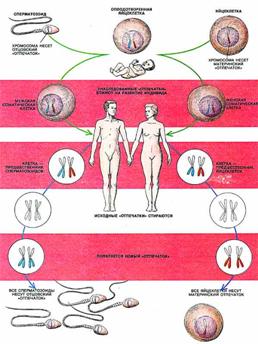
Рис. 2 Геномный импринтинг у человека
1.3 Болезни импринтинга
Проявления геномного импринтинга удивительным образом связаны с некоторыми заболеваниями человека. Неожиданно оказалось, что в природе уже существуют параллели тем экспериментальным состояниям, которые исследовали у мышей. Недавно Р. Николлс и его коллеги из Медицинской школы Гарвардского университета установили, что у многих больных с синдромов Прадера—Вилли обе хромосомы 15 унаследованы от матери (3).
Р. Николлс, Дж. Кнолл (тоже сотрудник Гарвардского университета) и Ч. Уильямс из Флоридского университета обнаружили связанную с геномным импринтингом закономерность у больных с синдромом Ангельмана. У таких больных нередко имеют место частичные делеции унаследованной от матери хромосомы 15, в результате чего полностью функциональна только отцовская хромосома 15.
Эти два заболевания, хотя и столь разные по клинической картине, могут быть связаны с различиями в импринтинге одних и тех же генов одной и той же хромосомы. Однако в отличие от ненормально крупных и мелких мышей, синдромы Прадера—Вилли и Ангельмана не удается представить просто как две стороны одной медали, т. е. объяснить избытком либо недостатком продуктов одних и тех же генов. Исследования Николлса и его коллег показали, что бывает очень трудно предсказать, каким образом конкретные признаки зависят от процесса геномного импринтинга. Полезно было бы изучить на этот предмет многие наследственные заболевания человека; не исключено, что найдутся указания на влияние геномного импринтинга (3).
Примером импринтинга целого генома у человека является истинный пузырный занос, который возникает при оплодотворении яйцеклетки, лишенной материнских хромосом, двумя сперматозоидами. Несмотря на наличие полноценного диплоидного набора, ранний эмбриогенез таких зигот протекает аномально: ткани собственно эмбриона вообще не формируются, однако бурно разрастается трофобласт. В случае двойного набора материнских хромосом развивается тератома — эмбриональная опухоль. Следовательно, у человека, как и у мыши, на ранних стадиях развития геном отца преимущественно обеспечивает развитие провизорных органов, а геном матери — эмбриональных структур. Только материнский или только отцовский геномы не в состоянии обеспечить нормальное развитие эмбриона (1, 3).
На организменном уровне эффект импринтинга обнаружен в связи с наличием в хромосомном наборе фрагментов или целых хромосом одного (материнского или отцовского) происхождения — так называемая однородытельская дисомия (ОРД), т.е. наблюдается качественный, а не количественный хромосомный дисбаланс. Известны два основных механизма образования ОРД у человека: коррекция трисомии до дисомии (гетеродисомия), происходящая в 1-м мейотическом делении, и коррекция моносомии до дисомии (изодисомия) — во 2-м мейотическом делении.
Феноменология импринтинга значительно лучше изучена у мыши, чем у человека, и поскольку известна гомология между хромосомами человека и мыши (примерно по 700 локусам), можно использовать данные по импринтингу, полученные на мышах, для целенаправленного поиска импринтинга по определенным локусам у человека.
Импринтированные гены и их транскрипты обнаружены на многих хромосомах человека — 1, 5, 6, 7, 11, 13, 15, 19, 20 и X. На хромосоме 7 мыши и хромосомах 1 и 15 человека найдены два больших кластера ортологичных импринтированных генов, т.е. эволюционно консервативных по статусу импринтинга. Идентифицированы гены с полиморфным импринтингом, т.е. с сочетанием моноаллельной экспрессии в одних тканях и диаллельной — в других. По-видимому, такая тканеспецифическая эпигенетическая модификация некоторых генов может быть одним из механизмов, обеспечивающих дифференциальную экспрессию генов клеток разных тканей в ходе развития (1).
В последние годы с помощью молекулярно-генетических методов феномен геномного импринтинга наблюдают и при мультифакториальных заболеваниях. Например, четко выраженный отцовский импринтинг обнаружен при атопическом дерматите, материнский — при бронхиальной астме и атопии у детей. При инсулинзависимом сахарном диабете выявлена более высокая вероятность отцовского импринтинга. Ген инсулина у человека расположен в кластере импринтированных генов 11р15 и гомологичен локусам в мышином геноме, подверженным импринтингу. Кроме того, обнаружена ОРД отцовского происхождения у детей с неонатальным сахарным диабетом.
Примеров заболеваний, в основе этиологии которых лежит нарушение функции импринтированных участков генома, довольно много, поэтому можно говорить об особом классе заболеваний человека — «болезнях импринтинга», которых насчитывается уже более 30. Основные из них приведены в табл. 1.
Таблица 1
Предполагаемые «болезни импринтинга» у человека
| Заболевание | Хромосома | Происхождение |
| Синдром Адамса—Оливера. | Материнское | |
| Болезнь Альцгеймера | Отцовское | |
| Синдром Энжельмена | 15 | Материнское |
| Атопия | 11 | То же |
| Церебеллярная атаксия | Отцовское | |
| Расщелина губы | То же | |
| Врожденный порок сердца | Материнское | |
| Семейные опухоли клубочков | 11 | Отцовское |
| Синдром ломкой хромосомы X | X | Материнское |
| Синдром Гольденхара | То же | |
| Хорея Гентингтона (ювенильная форма) | 4 | Отцовское |
| Идиопатический гипертрофический субаортальный стеноз | То же | |
| Злокачественная гипертермия | 19 | Материнское |
| Миотоническая дистрофия (врожденная) | 19 | То же |
| Нарколепсия | 6 | » » |
| Дефекты невральной трубки | Отцовское | |
| Нейрофиброматоз 1 | 17 | Материнское |
| Нейрофиброматоз II | 22 | То же |
| Поликистоз почек (два локуса) | 16 и ? | Материнское и отцовское |
| Поликистоз яичников | Материнское | |
| Синдром Прадера—Вилли | 15 | Отцовское |
| Псориаз | То же | |
| Псевдопсевдогипопаратиреоз | 20 | Материнское |
| Спиноцеребеллярная атаксия | Отцовское | |
| Туберозный склероз | Материнское | |
| Синдром Видемана—Беквита | 11 | То же |
| Билатеральная спорадическая ретинобластома | 13 | » » |
| Агенезия почек, аномалии лица | 16 | » » |
| Синдром лицевых аномалий, микрокрании, аномалий респираторного тракта, гепатомегалии | 14 | Отцовское |
| Синдром Сильвера—Рассела | 7 | Материнское |
| Синдром умственной отсталости, низкого роста, преждевременного полового созревания | 14 | То же |
| Заболевание | Хромосома | Происхождение |
| Синдром Адамса—Оливера. | Материнское | |
| Болезнь Альцгеймера | Отцовское | |
| Синдром Энжельмена | 15 | Материнское |
| Атопия | 11 | То же |
| Церебеллярная атаксия | Отцовское | |
| Расщелина губы | То же | |
| Врожденный порок сердца | Материнское |
микроделеция дисомия наследственный импринтинг