


Химия каренов
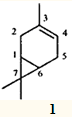
Одним из наиболее распространенных монотерпеновых углеводородов является 3-карен — 3,7,7-триметилбицикло(4.1.0)гепт-3-ен (1), входящий в состав многих эфирных масел и скипидаров.
 молекулярный радикальный перегруппировка карановый монотерпеноид
молекулярный радикальный перегруппировка карановый монотерпеноид
Интерес к 3-карену обусловлен наличием в его молекуле гем-диметилциклопропанового кольца — уникального фрагмента, являющегося важным структурным элементом ряда биологически активных соединений. Здесь можно упомянуть природные и синтетические пиретроиды, стеркуловую кислоту и ее эфиры, производные аминоциклопропанкарбоновой кислоты и др. Кроме того, 3-карен и родственные соединения, как и другие бициклические терпеноиды, способны к разнообразным скелетным перегруппировкам. Поэтому они — чрезвычайно перспективные модели для исследований в области фундаментальной органической химии, а также полезные исходные соединения для получения ценных продуктов — сесквитерпеноидов,3 феромонов,4 ювеиоидов,5 пиретроидов 6-7 и душистых веществ.7-9
Известные в настоящее время скелетные перегруппировки соединений ряда карана протекают с образованием структур нескольких типов: с бицикло(3.1.0)гексановым, бицикло(3.2.0)гептановым, п- и м-ментановым и 1,1,4-триметилциклогептановым скелетами. По механизмам перегруппировки классифицируются на ионные, радикальные и согласованные.
Существующие обзоры по карановым монотерпеноидам были написаны в основном более 25 лет тому назад и не включают скелетные перегруппировки, исследованные в последние десятилетия. Подробный обзор 10 посвящен перегруппировкам, протекающим по ионному механизму. В обзоре 11 в основном приведены перегруппировки карановых монотерпеноидов, в которых циклопропановое кольцо (ЦПК) раскрывается по внешним связям с образованием соединений ряда п- и м-ментана.
В обширной монографии Эрмана "Химия монотерпенов" 12 прекрасно изложены систематические данные по реакциям монотерпенов и рассмотрены механизмы этих реакций, однако фактический материал по превращениям терпеноидов ряда карана включает литературу до 1976 г. К настоящему времени получены новые результаты и появились новые представления о механизмах некоторых реакций, рассмотренных в этой монографии. Периодически публикуемые обзоры13-14 носят констатирующий, а не аналитический характер, поэтому механизмы описанных превращений в них как правило не рассматриваются и не обсуждаются. В последних обзорах, посвященных перегруппировкам терпеноидов, 3,15,16 превращения соединений с карановым скелетом практически не рассматриваются.
Целью курсовой работы является обобщение данных по скелетным превращениям терпеноидов каранового ряда. Более ранние работы привлечены в тех случаях, когда представляет интерес история вопроса или появилось новое понимание известных фактов. Перегруппировки рассмотрены в соответствии с классификацией по их механизмам.
II. Перегруппировки карановых монотерпеноидов, протекающие по ионному механизму
Реализация ионного механизма при молекулярных перегруппировках карановых монотерпеноидов обусловлена взаимодействием циклопропанового кольца с электронодефицитным карбениевым центром в α- или β-положении к циклу. Интерес к этому типу перегруппировок связан с возможностью образования интермедиатов с делокализованным зарядом. Согласно предложенной в обзоре10 классификации, ионные перегруппировки карановых структур подразделяются на гомоаллильную перегруппировку, перегруппировку с трансаннулярным участием ЦПК и перегруппировку типа Вагнера-Меервейна с сужением шестичленного цикла. Однако эта классификация не отражает в полном объеме разнообразие ионных перегруппировок карановых структур и является условной. Гомоаллильная перегруппировка — не единственная возможность скелетных превращений через карбокатион с делокализованным зарядом: в карановых структурах, содержащих двойные связи, может существовать и ион типа пентадиенильного, причем пути его образования могут быть различными для различных соединений. Кроме того, по нашему мнению, перегруппировку с трансаннулярным участием ЦПК и перегруппировку типа Вагнера - Меервейна более корректно рассматривать в ряду согласованных реакций, поскольку в них происходят алкильные сдвиги в заряженных системах.
1. Карбений-ионные перегруппировки
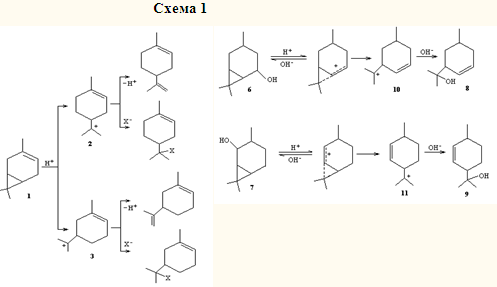
Молекула 3-карена содержит два реакционноспособных фрагмента: двойную связь и циклопропановое кольцо. Оба эти фрагмента характеризуются повышенной электронной плотностью, что делает молекулу реакционноспособной по отношению к электрофильным агентам. Хорошо известна перегруппировка 3-карена под действием кислот с образованием соединений ряда п- и м-ментана.12 Очевидно, что в этом случае циклопропановое кольцо выступает как изолированный реакционный центр, образуя карбениевые ионы 2 или 3. Двойная связь удалена и не взаимодействует с атомом углерода, несущим положительный заряд. Ионы стабилизируются путем выброса протона или захвата нуклеофила (схема 1).
Описанное превращение осуществляется в том случае, когда вначале протонируется ЦПК. Очевидно, что присоединение протона к двойной связи углеводорода 1 должно приводить к третичному иону 4.
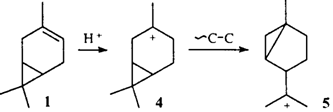
В ионах такого типа возможна перегруппировка, обусловленная так называемым "трансаннуляриым циклопропильным участием"10 (см. раздел IV.4), но ее результатом было бы образование иона 5 и продуктов иной структуры.
Причину того, что в случае 3-карена протежируется ЦПК, а не двойная связь, можно объяснить с позиций принципа жестких и мягких кислот и оснований (ЖМКО). Циклопропан — более жесткое основание, чем двойная связь (потенциалы ионизации 10.9 и 9.3 эВ соответственно). В молекуле 3-карена разница энергий высших занятых молекулярных орбиталей (ВЗМО) соответствующих фрагментов несколько меньше, но все еще достаточно велика (9.16 и 8.61 эВ).17 Жесткая кислота — протон — присоединяется вначале к жесткому основанию — ЦПК. Так, при взаимодействии эквимолярных количеств 3-карена и хлористого водорода образуются производные со скелетом п- и м-ментана.18 Сходство электронного строения двойной связи и ЦПК 19 обусловливает сходство их реакционной способности.20 Наличие катионоидного центра в α-положении к циклопропановому кольцу приводит к их сопряжению и образованию гомоаллильного иона. Направление реакции этого иона с нуклеофилом зависит от пространственных факторов, природы уходящей группы, степени сольватации молекулы (в реакциях сольволиза) и др.
X- — нуклеофил
Если атака гомоаллильного иона происходит со стороны ЦПК, это приводит к его раскрытию и в случае карановых производных — к перегруппировкам скелета.
Так, в условиях кислотного катализа каран-5-ол (6) и каран-2-ол (7) изомеризуются в м- (8) и п-ментен-8-олы (9) соответственно в результате атаки гидроксила по атому С(8) гомоаллильных ионов 10 и 11.21,22 Эти реакции можно рассматривать как региоспецифичные.
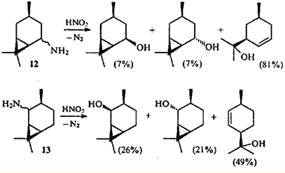
Подобным же образом протекает дезаминирование 5- (12)- и 2-аминокаранов (13), однако присутствие среди продуктов бициклических спиртов показывает, что в данном случае реакция не была доведена до конца.23
Гомоаллильные перегруппировки могут сочетаться с другими ионными перегруппировками. Так, при конденсации 3-карена с формальдегидом по Принсу среди продуктов реакции обнаружены два циклических эфира 14 и 15. Их образование объясняется последовательным протеканием перегруппировки типа Вагнера — Меервейна и гомоаллильной.24-25
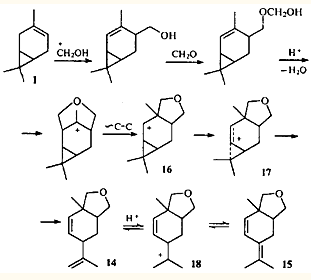
В данном случае перегруппировка Вагнера-Меервейна, протекающая как алкильный сдвиг, не приводит к изменению карановой структуры. Однако алкильный сдвиг возможен лишь потому, что в образующемся вторичном ионе 16 делокализация заряда с участием циклопропанового кольца приводит к его раскрытию и образованию гомоаллильного иона 17, обладающего меньшей энергией. Ион 17 стабилизируется путем выброса протона от одной из геминальных метильных групп, давая эфир 14. В кислой среде этот эфир изомеризуется в эфир 15 через классический катион 18.
2. Перегруппировки, протекающие через ионы с высокой степенью делокализации заряда
Очевидно, что в систему ЦПК - заряженный атом углерода может включаться одна или несколько двойных связей. Образующийся при этом гомопентадиенильный ион характеризуется большей степенью делокализации заряда, чем гомоаллильный, и имеет, соответственно, более низкую энергию. Поэтому реакции с участием гомопентадиенильных ионов протекают легче, чем с участием гомоаллильных. Изомеризация 4-карен-3-ола (19) в гидроксидиен 20 происходит при кипячении в воде даже в отсутствие кислотных катализаторов:26
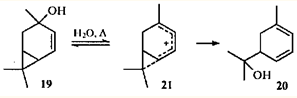
Как и при изомеризации соединений 6 и 7, необратимость атаки по циклопропановому фрагменту иона 21 обусловливает региоспецифичность реакции.
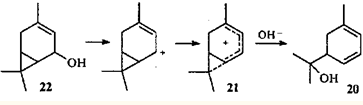
Следует отметить, что соседство (и, возможно, сопряжение) двойной связи и ЦПК в нейтральной молекуле не является обязательным условием образования гомопентадиенильного иона и, следовательно, протекание скелетной перегруппировки. С точки зрения механизма интересна селективная изомеризация кар-3-ен-5-ола (22) в м-мента-1,3-диен-8-ол (20): двойную связь и ЦПК объединяет в систему сопряжения возникающий между ними карбокатаонный центр.27
В систему сопряжения катионоидный центр - ЦПК может включаться не только соседняя двойная связь, но также и второй трехчленный карбоцикл. Участие такого интермедиата в реакции приводит к перегруппировке трициклооктанового скелета в замещенный циклогептадиен.28 Эта перегруппировка интересна тем, что в данном случае гем-дихлорциклопропановое кольцо раскрывается по внутренней связи, а гем-диметилциклопропановое — по внешней.
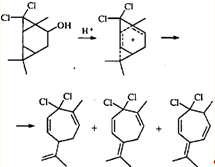
Из приведенных выше примеров ионных перегруппировок видно, что для гем-диметилциклопропанового кольца раскрытие по внешней связи является закономерностью, практически не знающей исключений. Трехчленный цикл, связанный с двумя атомами галогена, напротив, может вести себя по-разному. При метанолизе аддукта 3-карена с дибромкарбеном ЦПК раскрывается по двум направлениям, приводя к продуктам как с сохранением карановой структуры, так и к продуктам перегруппировки.29
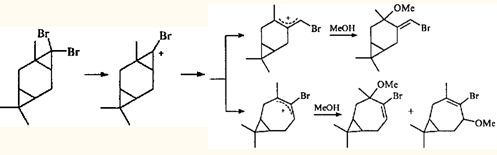
То, что трехчленные циклы в ионных реакциях раскрываются по разным связям, может быть обусловлено природой геминальных заместителей. гем-Диметильная группировка укорачивает (и усиливает) противолежащую связь циклопропана,30 а два геминальных атома хлора удлиняют и ослабляют ее. Геминальные атомы брома действуют таким же образом, но слабее.31
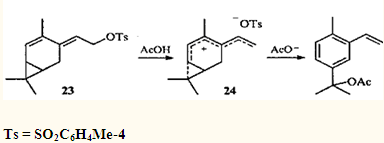
Чрезвычайно легко протекает ацетолиз тозилата 23;32 в данном случае интермедиатом является гомогептатриенильный ион 24, в котором степень делокализации заряда еще выше, чем в гомопентадиенильном.
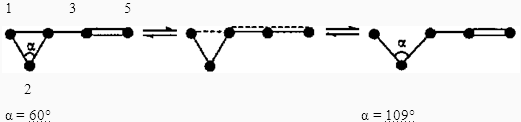
Выше было сказано, что гомоаллильные и гомопентадиенильные перегруппировки являются необратимыми, однако это утверждение не совсем корректно. Действительно, подобные превращения карановых производных можно провести со 100%-ной степенью конверсии. Это обусловлено геометрией иона и образующейся молекулы. Атом углерода ЦПК при С(2), не включенный в цепь сопряжения, в гомоаллильном ионе, в процессе его стабилизации меняет гибридизацию с sp2 на sp3 При этом угол С(1)—С(2)—С(3), равный в молекуле циклопропана 60°, в конечной молекуле увеличивается до 109°, что вызывает, в свою очередь, увеличение расстояния между атомами С(1) и С(3). Однако если эти атомы удастся каким-то образом сблизить, вновь становится возможным образование гомоаллильного иона.
На практике соединения с ментановым скелетом никогда не превращаются в карановые. Однако именно в карановых структурах, благодаря особенностям их строения, можно наблюдать ретро-гомоаллильную перегруппировку, при которой гомоаллильный ион образуется из линейного фрагмента молекулы и приводит к замыканию трехчленного цикла. Эффект подобного рода наблюдался при взаимодействии тозилата 25 с лчтийалюминийгидридом,33 триэтилалюминием,34 а также при его сольволизе.35
Очевидно, что в идеальном случае эта реакция должна протекать по механизму SN1. Однако скорее всего в реакции участвуют контактные ионные пары: происходит очень сильная поляризация субстрата с почти полным разделением зарядов, но образовавшийся катион стабилизирован противоионом. Отщепление тозилатной группы в соединении 25 приводит к катиону с зарядом на первичном атоме углерода, который гораздо менее стабилен, чем третичные ионы, образующиеся при протонировании ЦПК. Стабилизация иона может осуществляться путем сопряжения заряженного атома с двойной связью, причем перекрывание -орбиталей происходит не вдоль σ-связи, а через пространство ("through space").
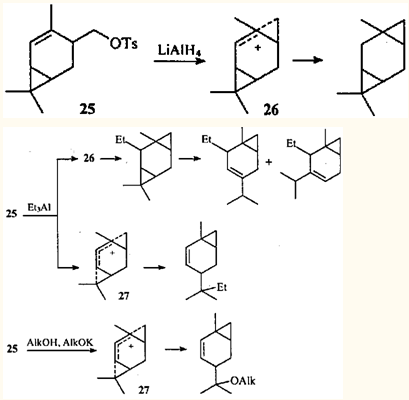
Сближению этих фрагментов способствует тесное сольватное окружение, поскольку процесс должен происходить быстро. Этот путь приводит к образованию гомоаллильного иона 26 и/или дигомопентадиенильного иона 27. В том случае, когда интермедиатом является ион 27, происходит совершенно необычная перегруппировка: фактически одна молекула претерпевает одновременно две перегруппировки различной направленности — гомоаллильную, приводящую к раскрытию циклопропанового кольца, и ретро-гомоаллильную, приводящую к замыканию нового трехчленного карбоцикла.
Тут уместно привести чрезвычайно интересный факт: 2-тозилоксиметил-3-карен (28) при ацетолизе дает тот же эфир 29, что и 4-тозилоксиметил-2-карен (25).36
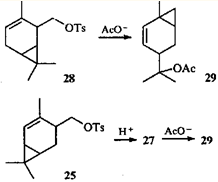
Механизм образования ацетата 29 пока не выяснен. Обращает внимание также отсутствие продуктов, образования которых можно было бы ожидать, опираясь на данные по превращениям эфира 25. Известно еще одно достаточно необычное превращение тозилата 28: при пиролизе он дает тот же углеводород, что и тозилат 25, а именно 1-метил-4-изопропенилбицикло(4.1.0)гепт-2-ен (30).36
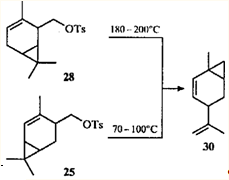
Механизм пиролиза эфира 2537 будет рассмотрен ниже, как и возможный механизм пиролиза тозилата 28.
Механизмы ионных перегруппировок, включающих ионы с делокализованным по цепи сопряжения зарядом, являются, конечно, гипотетическими, основанными на строении конечных продуктов. До сих пор неясно, действительно ли возможно сопряжение в таких системах. Ниже, при рассмотрении гидрирования 2-карена (31) будет показано, что соседство двойной связи и ЦПК отнюдь не гарантирует их сопряжения, по крайне мере в основном состоянии нейтральной молекулы. 17,38 Было бы чрезвычайно полезно проверить с помощью современных квантово-химических расчетов, возможна ли при перегруппировках соединений ряда карана делокализация заряда в системе сопряжения, в которой одним из элементов является ЦПК.
III. Перегруппировки, протекающие по радикальному механизму
Радикальные перегруппировки монотерпеноидов каранового ряда происходят, как правило, при фотохимических реакциях. Фотохимические превращения таких соединений достаточно подробно описаны в обзоре 12. Для 3-карена перегруппировки в условиях облучения не наблюдались. Известно лишь его фотохимическое окисление с образованием трех гидропероксидов.39 Изомерный 2-карен при облучении в присутствии сенсибилизатора перегруппировывается в 1,4,4-триметилбицикло(3.2.0)гепт-2-ен (32), который был использован для синтеза грандизола.4
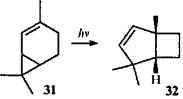
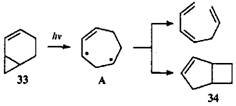
В данном случае механизм перегруппировки не рассматривался, но в литературе описан механизм для родственного соединения — 2-норкарена (33).40 При облучении соединения 33 основным продуктом был цис-гепта-1,3,6-триен, т.е. преобладал (2π+2ω+2σ)-процесс. Образование небольшого количества бицикло(3.2.0)гепт-2-ена (34) позволило авторам предложить общий дирадикальный интермедиат А с семичленным циклом:
Существует точка зрения,12,41 что образование соединений с бицикло(3.2.0)гептановым скелетом при облучении некоторых карановых терпеноидов может протекать как фотоиндуцированная сигматропная перегруппировка. В пользу этого предположения говорит и факт параллельного образования борненовой структуры из кетона 35.
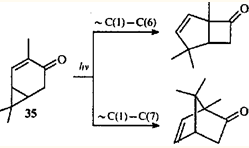
Косвенным подтверждением согласованного механизма перегруппировки кетона 35 можно считать результаты облучения 4-гидроксиметил-2-карена (36) ртутной лампой высокого давления:42 в присутствии сенсибилизатора (sens) образуются только бициклические стереоизомерные спирты 37 и 38, а при прямом облучении в гексане — еще и моноциклический спирт 39.
Кропп, исследовавший эту реакцию,42 проводил аналогию с 1,3-сопряженной системой, постулируя образование аллил-радикалов. При этом ЦПК может претерпевать гомолитический разрыв как "внутренней", так и одной из "внешних" связей. По-видимому, применение сенсибилизатора приводит к тому, что молекула превращается в дирадикал 40 из триплетного состояния: при этом рвется самая слабая связь ЦПК. В синглетном возбужденном состоянии молекула обладает большей энергией, что и приводит к разрыву обеих возможных связей ЦПК. В данном случае происходит разрыв связи С(1)—С(7), а не ее сдвиг. Очевидно, рекомбинация и образование новой связи между атомами С(1) и С(8) радикала 41 (что приводило бы к борнановому скелету) невозможны из-за большего расстояния между этими атомами, чем расстояние между атомами с неспаренными электронами дирадикала 40.
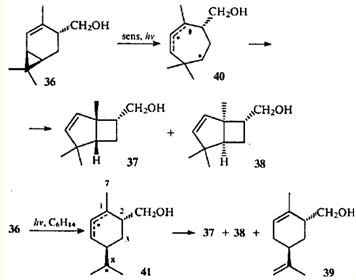
Облучение 4-ацетил-2-карена (42) в присутствии сенсибилизатора дает продукты с бицикло(3.2.0)гептановой структурой,43 т.е. замена гидроксиметильной группы на ацильную не влияет на механизм реакции.
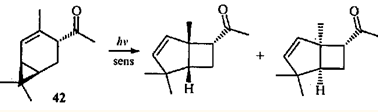
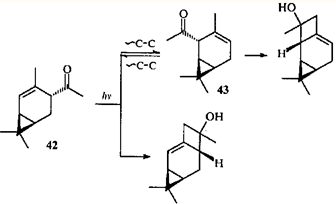
Однако облучение кетона 42 в отсутствие сенсибилизатора вызывает принципиально иную перегруппировку: образуется продукт 1,3-ацильного сдвига (2-ацетил-3-карен (43)), и трициклические спирты — продукты циклизации II типа по Норришу обоих присутствующих в реакционной среде кетонов.43
Не совсем обычно ведут себя при облучении цис- (44) и транс-4-караноны (45). Хотя строение молекул позволяет осуществить расщепление II типа по Норришу связей С(3)—С(4) и С(4)—С(5), среди продуктов фотопревращений цис-каранона (44) обнаружены только соединения, образовавшиеся в результате расщепления связи С(4)—С(5).44
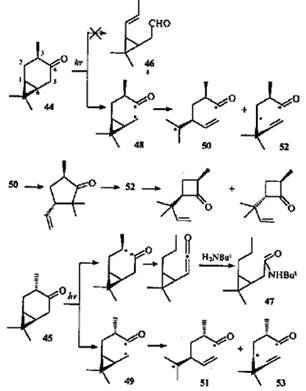
Вопреки ожиданиям, не наблюдалось образования альдегида 46 и эпимеризации кетона 45. Однако при облучении последнего в присутствии трет-бутиламина в продуктах реакции было найдено небольшое (>1%) количество амида 47. Это свидетельствует о том, что гомолитический разрыв связи С(3)—С(4) все-таки происходит, но в крайне незначительной степени.
Формально фотохимическое поведение кетонов 44 и 45 очень похоже на аллильные перегруппировки β,γ-ненасыщенных кетонов.45 Кропп с соавторами считал фотохимическую перегруппировку каранонов 44 и 45 первым примером циклопропильного аналога фотохимической аллильной перегруппировки.44 Однако в случае β,γ -ненасыщенных кетонов реакция обратима, а в случае кетонов 44 и 45 — нет.
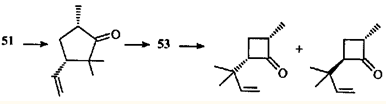
По аналогии с β,γ -ненасыщенными кетонами можно полагать, что направление α-расщепления углеродных связей обусловлено "аллильным" влиянием ЦПК, ослабляющим связь С(4)—С(5) за счет эффекта сопряжения или индуктивного. В образовавшихся радикалах 48 и 49 должны раскрыться связи С(1)—С(6) или С(1)—С(7). Соотношение присутствующих в продуктах реакций циклопентанонов и циклобутанонов (3:1) отражает преимущественное раскрытие более замещенной связи С(6)—С(7) и относительную стабильность третичного (50, 51) и вторичного (52, 53) радикалов.
Есть и еще одно отличие аллильных фотохимических перегруппировок β,γ-ненасыщенных кетонов от аналогичных циклопропильных. Аллильная перегруппировка считается согласованным процессом.45 Но превращение каранонов 44 и 45, по мнению авторов,44 не может быть согласованной реакцией, по крайней мере об этом свидетельствует образование эпимерных циклобутанонов.
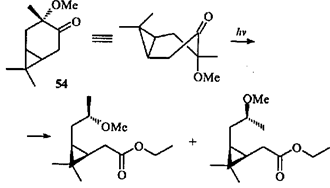
Направление скелетной фотохимической перегруппировки терпеноидов ряда карана зависит от ориентации заместителей: прямое облучение транс-3-метокси-4-каранона (54) в этаноле протекает с раскрытием шестичленного цикла по связи С(3)—С(4) (что для β,γ-циклопропилкетонов бывает крайне редко) и присоединением этанола.39
В случае цис-3-метокси-4-каранона (55) с экваториальной метоксигруппой наблюдается исключительно элиминирование II типа по Норришу и образование каранона 44; требуемое плоское шестичленное переходное состояние (ПС) А легко достижимо, и образование кетона 44 успешно конкурирует с расщеплением связи С(3)—С(4).39
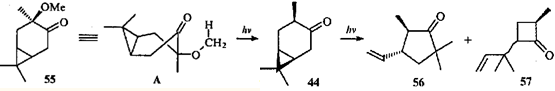
Кетон 44 под действием света подвергается гомолитическому разрыву связи С(4)—С(5) и гомоаллильной перегруппировке, характерной для β,γ -циклопропилкетопов.44,46 При этом образуются циклопентанон 56 и циклобутанон 57.
Таким образом, перегруппировки карановых монотерпеноидов под действием света можно разделить на две группы: перегруппировки, в которых в качестве хромофора выступают элементы углеродного скелета — циклопропан и двойная связь, — и перегруппировки, в которых хромофором является функциональный заместитель. Применение сенсибилизаторов приводит к перегруппировкам первого типа с образованием производных бицикло(3.2.0)гептана. Воздействие облучения в отсутствие сенсибилизатора, как правило, приводит к перегруппировкам второго типа, при этом возможны разрыв большого или малого циклов, сужение большого цикла или образование трициклической структуры. Направление реакции в каждом конкретном случае определяется природой и положением заместителя, являющегося хромофором.
IV. Согласованные реакции
Если ионным реакциям соединений каранового ряда посвящено множество работ, которые неоднократно систематизировались, то согласованные превращения значительно менее исследованы, и обобщение этого материала до сих пор не делалось. Справедливости ради следует отметить, что далеко не все реакции, которые формально относятся к согласованным, оказываются таковыми в действительности. Например, в случае 2,4-карадиена (58) 1,5-термический сдвиг формально является согласованной сигматропнои реакцией. Однако, во-первых, она протекает не через циклическое ПС (с одновременным разрывом и образованием связей С—С), а через дирадикальный интермедиат, а во-вторых, по сути, она не является сигматропным процессом. Иногда согласованный механизм осуществляется через сильно поляризованное ПС или сопряжен с ионным механизмом. Обзор подобных реакций карановых монотерпеноидов особенно интересен потому, что именно в них иногда осуществляются уникальные превращения, характерные только для этих соединений (например, перегруппировка Берсона-Вилькотта 47). Исследование согласованных реакций существенно расширяет наши представления о механизмах химических реакций вообще.
1. Интерконверсия норкарадиен - циклогептатриен
Одно из интереснейших скелетных превращений карановых структур — перегруппировки, связывающие терпеноиды ряда карана и ряда 1,1,4-триметилциклогептана, — привлекло внимание исследователей и привело к открытию такого замечательного явления в органической химии, как "быстрое" равновесие норкарадиен - циклогептатриен. Этой теме посвящено большое количество работ последней трети двадцатого века.47-55 Быстрое равновесие валентных таутомеров норкара-2,4-диена (59) и циклогепта-1,3,5-триена (60) обусловлено типично согласованной реакцией — дисротаторным раскрытием и замыканием трехчленного карбоцикла, причем этот процесс подчиняется правилам сохранения орбитальной симметрии, сформулированным Вудвордом и Гофманом.56
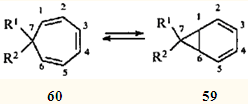
Для незамещенного углеводорода (R1 = R2 = H) это равновесие настолько сдвинуто в сторону моноциклического таутомера 60, что норкарадиен не удавалось обнаружить никакими физическими методами. Его существование постулировали на основании поведения замещенных аналогов (R1 = CN, R2 = CF3; R1 = CO2Me, R2 == Ph; R1 = CO2Me, R2 = C6H4OMe-4), которые в виде равновесных смесей с соответствующими циклогептатриенами обнаружены при исследовании последних методом низкотемпературного ЯМР.55 Только в 1981 г., четверть века спустя после появления гипотезы о валентной таутомерии циклогептатриена, удалось зафиксировать незамещенный норкарадиен при низкотемпературном (77 К.) фотолизе трицикло(3.2.2.02,4)нон-6-енди-8,9-она.57
Экспериментально установлено, что бициклическую структуру способны стабилизировать π-акцепторные заместители в положении 7.55 Природу такой стабилизации изящно объяснил Р.Гофман в рамках метода граничных МО.53 Очевидно, что метальные заместители стабилизаторами быть не могут, и поэтому для системы 2,4-карадиен (58) — 3,7,7-триметилциклогепта-1,3,5-триен (61) равновесие всегда сдвинуто в сторону моноциклического партнера. Однако эти валентные таутомеры — все-таки разные, реально существующие соединения; они обладают различной реакционной способностью, что должно проявляться (и проявляется, как будет показано ниже) в химических реакциях.
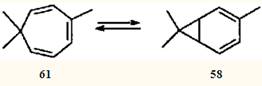
Перегруппировки, обусловленные описанной выше валентной таутомерией, будут обсуждены более подробно, чем ионные, поскольку в отечественной научной литературе проблема таутомерии норкарадиена практически не рассматривалась. К сожалению, даже исследователи, работающие в области химии терпеноидов, привлекают для объяснения расширения каранового скелета до циклогептанового недостаточно обоснованные гипотезы.12,58
Перегруппировка циклогептатриен — норкарадиен была обнаружена при изучении химических превращений эйкарвона (2,6,6-триметилциклогепта-2,4-диенона (62)) — терпеноида, впервые синтезированного Байером,59 но впоследствии обнаруженного в эфирных маслах различных растений.60
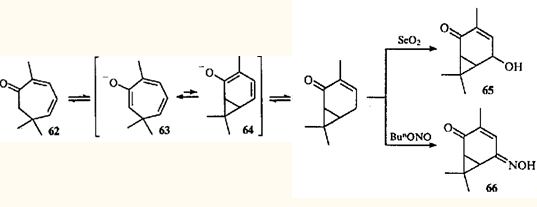
Гипотеза о валентной таутомерии моно- и бициклических енолов 63 и 64, как возможное объяснение скелетной перегруппировки кетона 62 при образовании 3,7,7-триметил-5-гидрокси-3-карен-2-она (65) и 3,7,7-триметил-5-оксиимино-3-карен-2-она (66), была выдвинута в 1956 г.61,62 Впоследствии оказалось, что гораздо более распространенными являются перегруппировки карановых производных в триметилциклогептановые, поскольку равновесие в динамическом взаимопревращении двух таутомеров сильно сдвинуто в сторону моноциклического партнера.63
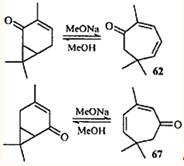
2,5,5-Триметилциклогепта-2,4-диенон (67), так же как и кетон 62. обнаружен в скипидарах из Pinus silvestris L.64 По-видимому, присутствие этих кетонов в живичном скипидаре объясняется тем, что из 3-карена (одного из основных компонентов скипидара) в процессе развития растения образуются непредельные кетоны ряда карана, которые затем согласованной электроциклической реакцией превращаются в моноциклические диеноны. Подтверждением этого могут служить результаты жидкофазного окисления 3-карена: содержание кетонов 62 и 67 в продуктах реакции достигало 20 и 28%.65
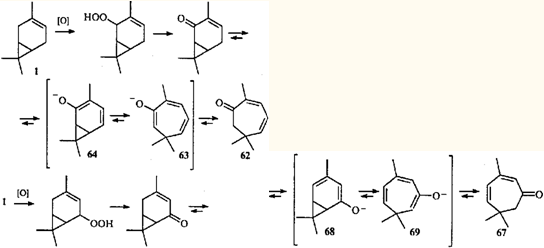
Следует отметить, что перегруппировки 64 →63 и 68 →69 — единственное на сегодняшний день объяснение присутствия в эфирных маслах кетонов с семичленным циклом. Общая схема биогенезиса терпенов 66,67 вообще не предусматривает образования подобных соединений. Однако широкое распространение в природе 3-карена и его производных и тот факт, что триметилциклогептановые соединения присутствуют только в тех эфирных маслах, где есть терпепоиды ряда карана, можно считать косвенным доказательством предположения о путях образования терпеноидов с семичленным циклом. Гораздо менее правдоподобным, по нашему мнению, является предположение о возможности образования эйкарвона (62) в процессе биосинтеза 12 тем же путем, каким его получал Байер: гидробромированием карвона и последующим дегидробромированием образовавшегося 8-бром-1-ментен-6-она.59
Наконец, серьезным аргументом в пользу перегруппировки 58 → 61 как причины образования циклогептановых терпеноидов не только invitro, но и invivo, служит обнаружение триена 61 в сосновом живичном скипидаре и в живицах отдельных деревьев Pinus silvestris.68 Поскольку этот триен существует только в паре с кара-2,4-диеном, который присутствует в скипидаре и живице, можно сделать вывод о возможности каталитического (ферментативного) дегидрирования 2-карена, также присутствующего в скипидарах.
Двойственная природа триена 61 чрезвычайно наглядно проявляется в реакции гидрирования (H2, Pt, 20°С); при неполном гидрировании в продуктах реакции наряду с триметилциклогептадиенами и триметилциклогептенами обнаруживаются также 3-карен (1) и 2-карен (31). Продукт исчерпывающего гидрирования представляет собой смесь 1,1,4-триметилциклогептана 70(96%) и карана 71 (4%).69
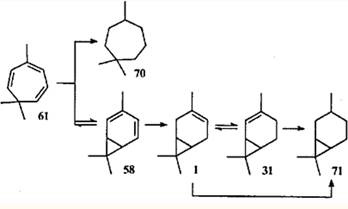
Разумеется, соотношение конечных продуктов гидрирования (70:71 = 96:4) не отражает содержания таутомеров в исходной равновесной смеси. Сравнительно большое количество карана 71 может быть обусловлено тем, что плоская диеновая система карадиена 58 успешно конкурирует с неплоскими диеновыми фрагментами триена 61 в реакции 1,4-присоединения водорода.
Необычным является гидрирование 3-карена. В 1966 г. Кокером с соавт.70 было установлено, что продукт гидрирования 3-карена на платине представляет собой смесь соединений 71 (98%) и 70 (2%), а при гидрировании на палладии преобладающим продуктом является углеводород 70 (74%).70 Позже оказалось, что гидрирование 3-карена на медно-никелевом катализаторе протекает с образованием исключительно 1,1,4-триметилциклогептана (70).58 Авторы обеих работ считали, что такое течение реакции обусловлено изомеризацией 3-карена (1) в 2-карен (31) и 1,4-присоединением водорода к винилциклопропановой системе последнего с образованием углеводорода с семичленным циклом.
При этом подразумевалось, что возможность или невозможность 1,4-присоединения, которое является ключевой стадией перегруппировки, зависит от природы катализатора. Но 1,4-присоединение возможно только в том случае, если имеется сопряжение в диеновом (или, для углеводорода 31, в гомодиеновом) фрагменте. Однако молекула терпена 31 находится в конформации "плоское кресло",38 что ставит под сомнение возможность такого сопряжения. Фотоэлектронный спектр показывает, что π-орбиталь и ВЗМО диметилциклопропанового кольца в молекуле соединения 31 практически не взаимодействуют.17 То что 1,4-присоединение для молекулы 2-карена (31) неосуществимо, подтверждено экспериментально: взаимодействие синглетного кислорода — исключительно активного диенофила — с этим терпеном приводит лишь к аллильным гидропероксидам,27 в то время как его реакции, например, с α-фелландреном и α-терпиненом,71 дают продукты 1,4-присоединения — эндопероксиды.
При обсуждении механизма реакции Кокер с соавт.70 не учитывал полученные им же результаты: при использовании палладиевого катализатора содержание углеводорода 70 увеличивалось с повышением температуры гидрирования и составляло 100% при температурах выше 70°С, т.е. на соотношение продуктов реакции прежде всего влияет температура. Аналогичная температурная зависимость позже была установлена и для гидрирования на платине: если при 20°С содержание углеводорода 70 составляет 2-3%, то при 70 и 90°С — 37 и 46% соответственно.72
Скелетная перегруппировка 3-карена при гидрировании становится объяснимой, если учесть, что и платина, и палладий катализируют как прямую, так и обратную реакции и способны осуществлять дегидрирование в присутствии свободного водорода.73 Гидрирование 3-карена в результате присоединения водорода к двойной связи приводит к карану; дегидрирование 2-карена, всегда имеющегося в реакционной среде (вследствие изомеризации 3-карена, которая происходит на катализаторе в присутствии водорода), обусловливает появление диена 58, триена 61 и других соединений с измененным скелетом.74 Повышение температуры благоприятствует эндотермической реакции дегидрирования и, соответственно, увеличению содержания перегруппированных продуктов. Следует отметить, что гидрирование на медно-никелевом катализаторе, когда углеводород 71 был единственным продуктом, также проводили при высокой температуре (180°С).58
Прямым доказательством реализации именно этого механизма служит диспропорционирование 2- и 3-каренов на палладии и платине в отсутствие свободного водорода. В этом случае среди продуктов был идентифицирован триен 71, что подтверждает предположение о дегидрировании 2-карена (31) как о стадии, отвечающей за образование перегруппированных продуктов.75 В реакционной смеси присутствовали также триметилциклогептадиены 72 и 73. Однако ни один из известных путей изомеризации терпенов 1 и 31 (в том числе и каталитический) не приводит к подобному расширению цикла.
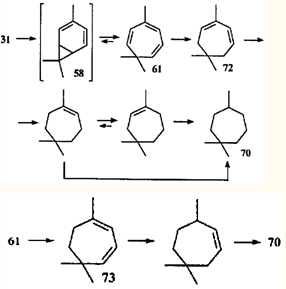
На основании сказанного выше можно сделать вывод, что перегруппировка карановых структур в триметилциклогептановые происходит в том случае, когда в ходе реакции возможно образование диена 58 (или его производных). Проще всего получить карадиен 58 из производных 2- и 4-карена, имеющих функциональную группу в аллильном положении. Так, при дегидрохлорировании 4-хлор-2-карена (74) образуется триен 61.76 В продуктах превращения эпоксида (75) в присутствии рениевых катализаторов обнаружили 4-карен-3-ол (76), который при дегидратации также дает углеводород 61.77
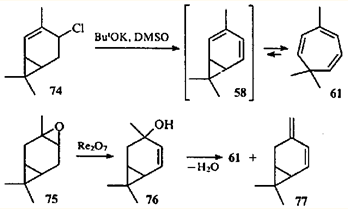
Теоретически дегидратация спирта 76 может идти и по другому направлению: с образованием терминальной метиленовой группы. Соответствующий углеводород — 3(10),4-карадиен (77) — действительно был обнаружен в продуктах реакции эпоксида 75.77 Углеводород 77, полученный впервые дегидрохлорированием 4-хлор-3(10)-карена (78),78 является первым устойчивым диеном с карановым скелетом.
Дегидратацией спирта 36 в присутствии гидроксида калия был синтезирован второй представитель этого ряда — 4-метил-3(10),4-карадиен (79).79
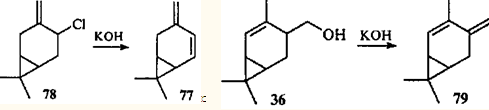
Диены 78 и 79 устойчивы (т.е. не склонны к таутомерным превращениям), так как в отличие от соединения 58 они имеют трансоидную диеновую систему, для которой невозможно электроциклическое замыкание и, следовательно, согласованная перегруппировка. Однако эти соединения весьма реакционноспособны: д