


Теоретические основы электрохимической коррозии
Металлы составляют одну из основ цивилизации на планете Земля. Их широкое внедрение в промышленное строительство и транспорт произошло на рубеже XVIII-XIX веков. В это время появился первый чугунный мост, спущено на воду первое судно, корпус которого был изготовлен из стали, созданы первые железные дороги. Начало практического использования человеком железа относят к IX веку до нашей эры. Именно в этот период человечество перешло из бронзового века в век железный.
Термин коррозия происходит от латинского слова «corrodere», что означает «разъедать , разрушать». Коррозия - это самопроизвольный процесс разрушения материалов и изделий из них под химическим воздействием окружающей среды. Коррозия металлов - разрушение металлов вследствие физико-химического воздействия внешней среды, при котором металл переходит в окисленное (ионное) состояние и теряет присущие ему свойства.
Особое место в комплексе мероприятий по обеспечению бесперебойной эксплуатации оборудования отводится надежной защите его от коррозии и применению в связи с этим высококачественных химически стойких материалов.
Целью курсового проекта является изучение теоретических основ электрохимической коррозии. Для ее достижения были поставлены следующие задачи: дать общую характеристику и классифицировать процессы коррозии, выявить условия возникновения коррозиционного процесса, изучить основы кинетической теории коррозии и ее приложение к коррозии идеально чистых металлов, коррозию технических металлов и методы защиты.
1 Общая характеристика процессов коррозии
Коррозия металлов определяется как процесс (и как результат) самопроизвольного разрушения металлов при их химическом, электрохимическом или биохимическом взаимодействии с окружающей средой. Коррозия представляет собой нежелательный и непреднамеренный процесс. Электрохимическое растворение анодов из черновой меди в ванне по ее рафинированию нельзя считать коррозионным процессом, так как оно является необходимым и желательным звеном в очистке меди от примесей. В то же время электрохимическое растворение железного анода в ванне по электролизу воды следует отнести к категории коррозионных процессов, поскольку оно здесь нежелательно. Разъедание стенок железной цистерны при перевозке в ней серной кислоты считается коррозионным разрушением, а растворение железа в серной кислоте с целью получения чистого реактивного сульфата железа не принято рассматривать как его коррозию, хотя в основе обоих процессов лежат одни и те же явления.
Коррозия - это не только нежелательный, но и самопроизвольный процесс, и в подавляющем большинстве случаев ее протекание не связано с подведением энергии от какого-либо внешнего ее источника. Эту особенность коррозии легко понять, если учесть, что корродируют обычно металлы (черные и цветные), встречающиеся в природе не в самородном состоянии, а как соответствующие минералы и руды. На извлечение этих металлов из руд или минералов расходуется большое количество энергии. Эти же металлы переходят в результате их коррозионного разрушения снова в окислы, сульфаты, карбонаты и другие соединения, в форме которых они обычно встречаются в природе. Процесс коррозии, поскольку он приводит к регенерации исходных соединений, термодинамически более устойчивых по сравнению с чистыми металлами, протекает с уменьшением свободной энергии и совершается самопроизвольно. Напротив, металлы, которые в природе обычно встречаются в чистом виде (золото, платина и другие), не корродируют, если только условия их использования не слишком отличаются от природных. Неудивительно поэтому, что разрушение многих металлов проходит со значительной скоростью и приносит колоссальный ущерб всем отраслям народного хозяйства (1).
2 Классификация процессов коррозии
Различают химическую, биохимическую и электрохимическую коррозию металлов. Химическая коррозия металлов представляет собой их самопроизвольное разрушение, в основе которого лежат законы обычных гетерогенных химических реакций. Разрушение металлов под действием агрессивных газов при высоких температурах, исключающих конденсацию влаги на поверхности металла, а также, по-видимому, их растворение в условиях контакта с органическими средами, не проводящими тока, относятся к процессам химической коррозии. Биохимическая коррозия, или биокоррозия, вызывается жизнедеятельностью различных микроорганизмов или использующих металл как питательную среду, или выделяющих продукты, действующие разрушающе на металл. Биокоррозия обычно накладывается на другие виды коррозии. Для ее развития наиболее благоприятны почвы определенных составов, застойные воды и некоторые органические продукты.
Электрохимическая коррозия подчиняется законам электрохимической кинетики. Скорость ее можно определить на основе законов Фарадея. Электрохимическая коррозия встречается чаще всего и наиболее опасна для металлов. Она может протекать в газовой атмосфере, когда на поверхности металла возможна конденсация влаги (атмосферная коррозия), в почвах (почвенная коррозия) и в любых растворах электролитов (жидкостная коррозия).
Особым случаем электрохимической коррозии следует считать электрокоррозию - коррозию за счет внешнего электрического тока. К электрокоррозии, кроме разрушения нерастворимых анодов, относятся коррозия трубопроводов с токопроводящими жидкостями, а также растворение стенок электролитических ванн и подземных металлических сооружений под действием ответвленного постоянного тока (коррозия блуждающими токами). Один из участков металлического сооружения принимает ток (катодный участок) от какого-либо внешнего источника электрической энергии, а на другом - ток переходит в окружающую ионнопроводящую среду (анодный участок); разрушается при этом анодный участок.
В зависимости от характера разрушений, сопровождающих процесс электрохимической коррозии, различают сплошную коррозию, захватывающую всю поверхность металла, и местную, локализующуюся на определенных участках. Металлы, в зависимости от скорости их коррозии в данной среде, разделяют на устойчивые и неустойчивые. По тому, с какой скоростью разрушается металл в различных средах, их определяют как агрессивные или неагрессивные в коррозионном отношении. Для оценки коррозионной устойчивости металлов и агрессивности сред были предложены различные условные шкалы. Скорость коррозии выражают несколькими способами. Наиболее часто пользуются весовым и токовым показателями коррозии. Первый из них дает потерю веса (в граммах или килограммах) за единицу времени (секунду, час, сутки, год), отнесенную к единице площади (квадратный сантиметр, квадратный метр) испытуемого образца. Во втором случае скорость коррозии выражается силой тока (в амперах или миллиамперах), приходящейся на единицу площади образца (1,2).
3 Условия возникновения коррозионного процесса
Коррозия металлов представляет собой частный случай неравновесных электродных процессов; в то же время ей свойственны некоторые особенности, отличающие ее от других неравновесных электродных процессов. Для протекания коррозионного процесса совсем не обязательно наложение внешнего тока и тем не менее растворение металла в условиях коррозии совершается со скоростями, сравнимыми с теми, какие наблюдаются при растворении металлических анодов в промышленных электролизерах.
Если кусок какого-либо металла М приведен в контакт с водным раствором его соли МА, то через некоторое время на границе между металлом и раствором устанавливается значение потенциала, которое в дальнейшем будет сохраняться почти неизменным. Эта постоянная, или почти постоянная, величина может отвечать установившемуся равновесию между металлом и раствором или стационарности электродного процесса. Какой из этих двух случаев реализуется в действительности, определяется, в первую очередь, самой величиной электродного потенциала. Если термодинамический электродный потенциал металла имеет величину, при которой в данных конкретных условиях исключено протекание всех других процессов, (кроме обмена металлическими ионами между металлом и раствором), то установившаяся величина потенциала отвечает его равновесному значению в данных условиях. Скорость переходов ионов металла в двух противоположных направлениях выравнивается при достижении состояния равновесия и становится равной току обмена (уравнение 1 ), а установившееся значение потенциала отвечает его термодинамической величине.
![]() (1)
(1)
Примером таких систем может служить серебро, опущенное в раствор нитрата серебра.
Положение существенно меняется, если термодинамический электродный потенциал металла имеет величину, при которой наряду с ионизацией и разрядом ионов металла возможен хотя бы один дополнительный электродный процесс. В этом случае заряды через границу раздела между металлом и раствором переносятся уже не одним, а двумя сортами частиц. Установившееся постоянное значение потенциала не обязательно свидетельствует о достижении равновесного состояния. Оно указывает лишь на то, что суммарное число зарядов, переходящих через границу в одном направлении, равно суммарному числу зарядов, пересекающих ее в обратном направлении (уравнение 2):
![]() (2)
(2)
Если предположить, что дополнительным электродным процессом будет выделение и ионизация водорода, так называемая коррозия с водородной деполяризацией, то вместо уравнения 2 можно написать уравнение 3:
![]() , (3)
, (3)
где индекс М относится к металлу, а индекс Н — к водороду.
Когда скорости всех частных процессов сравнимы и ни одной из них в уравнении 3 пренебречь нельзя, тогда установившаяся величина потенциала не отвечает ни потенциалу металлического электрода первого рода (или металлического электрода второго рода, что возможно, если металл покрыт слоем его труднорастворимого соединения), ни потенциалу водородного электрода. Это будет некоторая компромиссная величина, зависящая от соотношения скоростей всех частных реакций (рисунок 1).
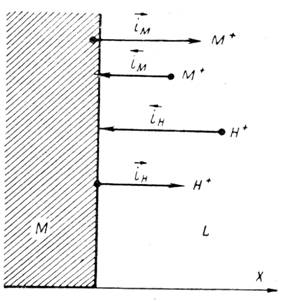
Рисунок 1 - Схема установления компромиссного или стационарного коррозионного потенциала при сравнимых скоростях катодной и анодной реакций:
Однако могут быть такие случаи, когда потенциал данного электрода не очень сильно отличается или от потенциала водородного электрода, или от потенциала соответствующего металлического электрода. Действительно, хотя частные токи в уравнении 3 не отвечают токам обмена в состоянии равновесия, они все же должны изменяться параллельно с величинами последних. Поэтому, если ток обмена металла i0M значительно больше тока обмена водорода i0Н, то с известным приближением можно допустить, что
![]() и
и ![]() .
.
Тогда вместо уравнения 3 можно написать в виде уравнения 4:
![]() (4)
(4)
Следовательно, поведение корродирующего электрода отвечает поведению обратимого металлического электрода, а установившееся значение компромиссного потенциала близко к равновесному потенциалу соответствующего металлического электрода и должно изменяться с концентрацией ионов металла в соответствии с формулой Нернста.
Изменение рН раствора не влияет при этом заметно на величину стационарного потенциала. Таким образом, здесь стационарный потенциал коррозии сε приводится к обратимому потенциалу металла Мεr (уравнение 5):
сε ≈ Мεr(5)
В качестве примеров таких систем можно привести разложение амальгам и электрохимическое растворение цинка.
Напротив, если ток обмена водорода значительно больше тока обмена металла, то
![]() ,
,
![]()
и, с некоторым приближением, можно получить уравнение 6:
![]() (6)
(6)
Теперь уже компромиссный потенциал близок к потенциалу водородного электрода в данных условиях. Его величина закономерно изменяется с рН и почти не зависит от концентрации ионов металла. Таким образом, здесь стационарный потенциал приводится к потенциалу водородного электрода (уравнение 7):
сε = Нεr (7)
Коррозия железа в слабокислых растворах служит примером подобного рода систем.
Необходимым условием электрохимической коррозии является совмещение на поверхности корродирующего металла реакций ионизации и разряда его ионов с какой-либо другой электродной реакцией, протекающей преимущественно в катодном направлении.
В водных средах, не содержащих иных окислителей, кроме ионов водорода и растворенного кислорода, это условие выполняется в том случае, если равновесный потенциал металла отрицательнее равновесного потенциала водородного или кислородного электродов в растворе данного состава. Добавление к раствору других окислителей с более положительным равновесным потенциалом увеличивает число металлов, способных корродировать. Так, серебро, стойкое в водных растворах не восстанавливающихся кислот, корродирует при добавлении окислителя с потенциалом более положительным, чем равновесный потенциал серебряного электрода.
4 Основы кинетической теории коррозии и ее приложение к коррозии идеально чистых металлов
Протекание процесса растворения металла указывает на то, что в данных условиях скорость ионизации больше скорости разряда его ионов:
![]()
В отсутствии внешнего тока и при условии сохранения неизменной величины потенциала такое соотношение скоростей возможно лишь в том случае, если скорость разряда водородных ионов на ту же величину больше скорости обратного процесса, то есть:
Скорость коррозии iс можно представить как разность между скоростью ионизации и скоростью разряда металлических ионов (уравнение 8) :
![]() (8)
(8)
Поскольку для процесса коррозии справедливо уравнение 3, то вместо уравнения 8 можно написать уравнение 9 и определить скорость коррозии как разницу между скоростями разряда ионов водорода и ионизации его молекул.
![]() (9)
(9)
Это позволяет вычислить скорость коррозионного разрушения металла по данным, относящимся к кинетике реакции выделения и ионизации водорода в коррозионных условиях (1,2).
5 Коррозия технических металлов
Все высказанные до сих пор соображения о коррозионном процессе относились к случаю электрохимического растворения идеально чистого металла с совершенно однородной по своим свойствам поверхностью. В реальных условиях коррозии подвергаются чаще всего технические металлы, содержащие примеси других металлов и неметаллических веществ. На их поверхности всегда можно обнаружить включения посторонних металлов. Кроме того, она обычно покрыта продуктами взаимодействия металла с окружающей средой, всевозможными загрязнениями. Естественно, что неоднородность поверхности и, прежде всего, присутствие на ней посторонних металлов с другими электрохимическими свойствами должны влиять на скорость коррозиии на характер ее протекания. Рассмотрим, например, как при этих условиях изменится скорость и характер коррозии цинка. Предположим, что в образцах цинка имеются включения или свинца, или серебра, или железа - обычные примеси в техническом цинке. Как следует из величин стандартных потенциалов, все эти металлы электроположительнее цинка. Поэтому можно предположить, что коррозии подвергается лишь цинк, а металлы-примеси остаются в неизменном виде. При этих допущениях коррозия должна протекать с водородной деполяризацией и ее скорость определяется кинетикой выделения водорода на корродирующем металле. В отличие от идеально чистого цинка, водород в этом случае может разряжаться не только на нем, но и на металле-примеси. Суммарная скорость выделения водорода, а следовательно, и суммарная скорость растворения цинка определяется поэтому кинетикой выделения водорода на основном металле и на включениях постороннего металла.
Таким образом, присутствие в цинке примесей свинца - металла с более высоким перенапряжением водорода - не увеличивает, а несколько снижает скорость коррозии.
Присутствие серебра в цинке должно увеличить скорость его коррозии. При выбранных условиях эта скорость возрастает в три с половиной раза. Однако увеличение скорости растворения не является единственным результатом загрязнения цинка серебром. Меняется и характер коррозии.
Серебро, обладая электроположительным потенциалом, не будет растворяться; на нем возможен лишь катодный процесс - выделение водорода. Цинк в присутствии включений серебра играет роль анода, и на нем сосредоточен весь процесс растворения. Кроме того, цинк выступает и в роли катода, обеспечивая выделение одной четвертой части от общего количества разряжающегося водорода.
Еще большего увеличения скорости коррозии и еще более полного разделения поверхности металла на анодные и катодные участки следует ожидать, когда цинк загрязнен железом. В этом случае анодные и катодные реакции оказываются пространственно разделенными. Анодная реакция целиком сосредоточена на поверхности цинка, а катодная - на поверхности включений железа.
В этих условиях корродирующий технический цинк можно рассматривать как совокупность гальванических микроэлементов, в каждом из которых железо (катод) является положительным полюсом, а анодно растворяющийся цинк - отрицательным. Коррозию такого технического металла можно на этом основании рассматривать как результат действия локальных гальванических элементов.
По теории местных элементов скорость коррозии (или пропорциональный ей электрический ток, возникающий в результате работы локальных гальванических пар) зависит не только от электрохимических свойств электродов этих пар, но и от омического сопротивления той среды, в которой совершается процесс коррозии и которая отделяет анод от катода. Определяющие скорость коррозии соотношения удобнее выразить графически при помощи так называемых коррозионных диаграмм. На коррозионной диаграмме (рисунок 2) потенциалы анода и катода или потенциалы анодного и катодного процессов представлены как функция силы тока.
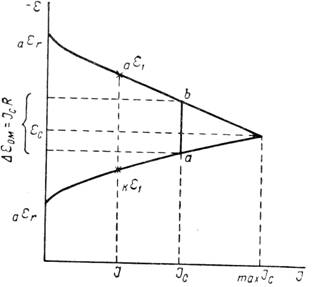
Рисунок 2 - Коррозионная диаграмма
Когда нет коррозионного процесса и сила тока равна нулю, тогда начальные значения потенциалов на аноде и катоде должны отвечать обратимым потенциалам анодной аεr и катодной kεr реакций в заданных условиях. За счет разности потенциалов анода и катода в системе появляется ток. При некоторой силе тока I потенциал анода сместится вследствие поляризации в сторону более положительных, а потенциал катода - в сторону более отрицательных значений. Пусть эти величины будут равны соответственно аε1 и kε1. Разность этих потенциалов под током меньше разности их обратимых потенциалов. С увеличением силы тока разность между потенциалом анода и катода будет непрерывно сокращаться. В пределе она окажется равной нулю, и поверхность корродирующего образца сделается эквипотенциальной. Здесь будет достигнута сила тока, отвечающая максимально возможной в данных условиях скорости коррозии maxIc, а потенциал образца станет равным величине εс , лежащей между аεr и кεr. Это максимальное значение силы тока может быть реализовано лишь тогда, когда сопротивление системы или равно нулю, или ничтожно мало. В противном случае, если омическое падение напряжения не равно нулю, скорость коррозии будет не maxIc , а некоторой меньшей величиной Iс. В этих условиях омическое падение напряжения ∆εOM численно равно длине отрезка ab. Потенциал анода в процессе коррозии будет отрицательнее потенциала катода на величину ∆εOM. Таким образом, скорость коррозии является функцией разности обратимых потенциалов анодной и катодной реакций, их поляризуемости и омического сопротивления коррозионной среды. Влияние каждого из этих факторов на скорость коррозии показано графически на рисунке 3 при помощи упрощенных коррозионных диаграмм. Скорость коррозии уменьшается, если при заданном сопротивлении и неизменной поляризуемости электродов обратимые потенциалы анодной и катодной реакций сближаются (рисунок 3,а), то есть Iс изменяется параллельно с величиной (кεr - аεr). Скорость коррозии становится меньше при увеличении общего сопротивления в системе корродирующий металл - коррозионная среда (рисунок 3,б). Повышение анодной (рисунок 3,в) или катодной (рисунок 3,г) поляризации также уменьшает скорость коррозии. Повышение поляризации может быть результатом появления дополнительных торможений как анодного, так и катодного процессов, либо уменьшением площади анодных или катодных участков. Сокращение площади данного электрода при неизменной силе тока увеличивает на нем плотность тока, а следовательно, и поляризацию.
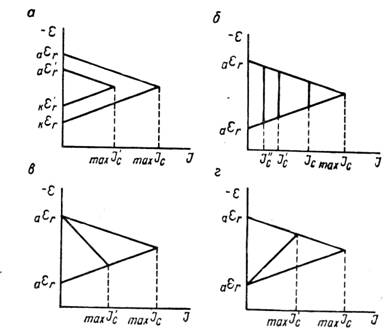
Рисунок 3 - Коррозионные диаграммы влияния различных факторов на скорость коррозии
В теории локальных элементов первоначально предполагалось, что катодный и анодный процессы должны быть обязательно пространственно разделены, и каждый из них может протекать лишь на вполне определенном участке поверхности корродирующего металла. На этом основании считалось, что идеально чистые металлы с совершенно однородной поверхностью не должны подвергаться коррозии. Такое заключение ошибочно и с термодинамической, и с кинетической точек зрения. Чтобы обеспечить протекание коррозии, необходима разница между обратимым потенциалом металла и потенциалами анодной и катодной реакций, возможных в данных условиях, а не пространственное разделение катодных и анодных участков. Анодные и катодные реакции в зависимости от степени однородности границы раздела металл - среда могут протекать на одной и той же поверхности или на ее различных участках. Совмещение катодных и анодных реакций типично для коррозии чистых металлов и амальгам; их более или менее полное пространственное разделение - для коррозии технических металлов. Меньшая стойкость технических металлов по сравнению с чистыми, а также изменение характера коррозионных разрушений во многом связаны с деятельностью гальванических микроэлементов основной металл - включение.
Коррозионные диаграммы, построенные на основе представлений теории локальных элементов, удобны для качественного рассмотрения процесса коррозии и для оценки возможного влияния на него различных факторов. В то же время их использование при количественных расчетах скорости коррозии связано со значительными трудностями. Скорость коррозии определяется изменением веса образца за единицу времени, отнесенного к единице его поверхности, или (в электрических единицах) плотностью тока i. Коррозионные же диаграммы, приведенные на рисунке 2 и 3, построены в координатах потенциал - сила тока (не включают в себя величины плотности тока, непосредственно характеризующей скорость коррозии).
Для ее расчетов нужны поэтому дополнительные данные. Задачу определения скорости коррозии можно проще решить на основе кинетической теории коррозии. В этом случае катодную и анодную поляризационные кривые снимают непосредственно на образце, коррозию которого изучают. Общую скорость коррозии выражают силой тока, отнесенной к единице всей поверхности металла, без разделения ее на катодные и анодные участки. При стационарном потенциале скорость коррозии, выражаемая силой тока анодного растворения металла, отнесенная ко всей его поверхности (включая и катодные зоны) должна быть равна скорости катодного процесса, например скорости выделения водорода. Последняя в случае снятия катодной поляризационной кривой будет равна силе тока, деленной на всю поверхность образца, включая анодные участки. Таким образом, при стационарном потенциале плотности тока для анодного и катодного процессов при указанном способе снятия поляризационных кривых должны быть одинаковыми. При этом предполагают, что омическими потерями можно пренебречь и, следовательно, рассматривать поверхность корродирующего металла как эквипотенциальную. Характер совмещенных поляризационных кривых, получаемых по этому методу, который показан на рисунке 4 (сплошные линии).
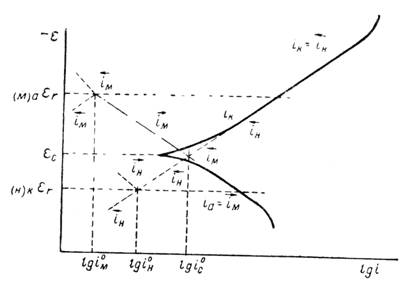
Рисунок 4 - Поляризационная диаграмма коррозионного процесса, протекающего с водородной деполяризацией
Точка пересечения анодной и катодной поляризационных кривых дает на оси абсцисс скорость коррозии, а на оси ординат - величину стационарного потенциала. Так как вблизи стационарного потенциала поляризационные данные перестают укладываться в полулогарифмическую зависимость, то скорость коррозии находят обычно по точке пересечения экстраполированных прямолинейных участков поляризационных кривых (пунктирные линии на рисунке 4). Сопоставление величин скорости коррозии, рассчитанных на основании поляризационных измерений, с полученными непосредственно из убыли веса (или по объему выделившегося водорода в кислых средах) для свинца, никеля и железа показало, что оба ряда данных совпадают друг с другом в пределах ошибок опыта. Это позволило широко использовать метод поляризационных измерений при количественном изучении коррозионных процессов.
Стационарный коррозионный потенциал εс лежит обычно между обратимыми потенциалами анодного и катодного процессов, обусловливающих появление коррозии. Он всегда положительнее равновесного потенциала анодной реакции и отрицательнее равновесного потенциала катодной реакции. Поэтому при стационарном потенциале скорость ионизации ![]() металла больше скорости разряда металлических ионов
металла больше скорости разряда металлических ионов ![]() , а скорость разряда иона водорода
, а скорость разряда иона водорода ![]() больше скорости ионизации его молекул
больше скорости ионизации его молекул ![]() . Такое соотношение скоростей сохраняется до тех пор, пока не будут достигнуты соответствующие равновесные потенциалы. В этом случае частные токи для каждого из двух процессов сделаются равными соответствующим токам обмена i0М и i0H. Продолжение катодной и анодной поляризационных кривых от стационарного потенциала до обратимых потенциалов электродных реакций показано на рисунке 4 штрихпунктирными прямыми. Скорость коррозии является функцией тока обмена катодной и анодной реакций. С увеличением тока обмена (при тех же равновесных потенциалах) скорость коррозии растет. Скорость коррозии должна меняться параллельно с изменением коэффициента переноса α. Таким образом, коррозионная диаграмма позволяет связать скорость коррозии с основными кинетическими параметрами лежащих в ее основе электродных реакций. Эту связь можно выразить и аналитически. При стационарном потенциале скорость коррозии должна быть равна скорости растворения металла и в то же время скорости катодной реакции; в рассматриваемом случае - скорости катодного выделения водорода. На этом основании можно написать следующее уравнение 10:
. Такое соотношение скоростей сохраняется до тех пор, пока не будут достигнуты соответствующие равновесные потенциалы. В этом случае частные токи для каждого из двух процессов сделаются равными соответствующим токам обмена i0М и i0H. Продолжение катодной и анодной поляризационных кривых от стационарного потенциала до обратимых потенциалов электродных реакций показано на рисунке 4 штрихпунктирными прямыми. Скорость коррозии является функцией тока обмена катодной и анодной реакций. С увеличением тока обмена (при тех же равновесных потенциалах) скорость коррозии растет. Скорость коррозии должна меняться параллельно с изменением коэффициента переноса α. Таким образом, коррозионная диаграмма позволяет связать скорость коррозии с основными кинетическими параметрами лежащих в ее основе электродных реакций. Эту связь можно выразить и аналитически. При стационарном потенциале скорость коррозии должна быть равна скорости растворения металла и в то же время скорости катодной реакции; в рассматриваемом случае - скорости катодного выделения водорода. На этом основании можно написать следующее уравнение 10:
ic = i0H℮-(kηH(1 – α)F)/RT = i0M℮(aηMα2zF)/RT , (10)
где kηH и aηM - соответственно катодная поляризация при выделении водорода и анодная поляризация при растворении металла;
(1— α) и α2 — коэффициенты переноса для реакции выделения водорода и растворения металла;
z — валентность металла.
Обозначим (1—α) через α2 и снимем индексы к и а у величины поляризации. Водородное перенапряжение в условиях коррозии равно разности между стационарным коррозионным потенциалом и обратимым потенциалом водородного электрода в данных условиях (уравнение 11):
ηH = εc - Hεr(11)
Аналогичное уравнение справедливо для анодной поляризации металла (уравнение 12).
ηМ = εc - Мεr(12)
Подставив в уравнение 10 вместо ηH и ηМ их значение из уравнения 11 и 12, решим полученное уравнение относительно стационарного потенциала коррозии (уравнение 13).
εc= 2.303 ![]() (13)
(13)
Подстановка этого значения εc в уравнение 10 после несложных преобразований приводит к уравнению 14.
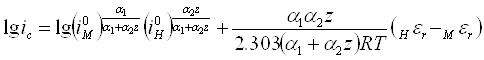 (14)
(14)
Уравнения 13 и 14 позволяют рассчитать величину потенциала металла в условиях его коррозии, а также скорость коррозионного разрушения, если только известны токи обмена, коэффициенты переноса и равновесные потенциалы анодной и катодной реакций.
Поляризационная диаграмма на рисунке 5, так же как и уравнения 13 и 14, относятся к тому случаю, когда скорость коррозии определяется чисто кинетическими ограничениями, то есть химической поляризацией. Это отвечает коррозии с водородной деполяризацией.
Другим важным случаем электрохимического разрушения металлов является их коррозия с кислородной деполяризацией. В связи с малой растворимостью кислорода в водных средах, а также в связи с тем, что его коэффициент диффузии значительно меньше коэффициента диффузии ионов водорода, скорость коррозии с кислородной деполяризацией обычно определяется диффузией. На рисунке 5 в упрощенном виде представлена типичная поляризационная диаграмма процесса коррозии с кислородной деполяризацией.
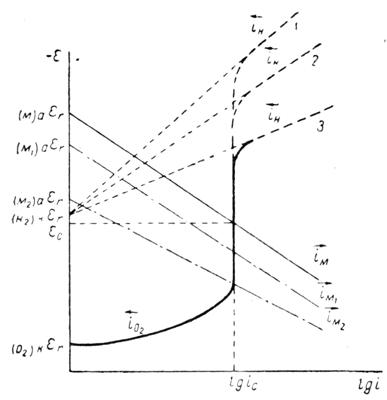
Рисунок 5 - Упрощенная поляризационная диаграмма процесса коррозии с кислородной деполяризацией
Скорость коррозии в этом случае оказывается равной предельному току диффузии кислорода к поверхности корродирующего металла (уравнение 15).
ic= 2oid(15)
Скорость коррозии с кислородной деполяризацией поэтому почти не зависит (в известных пределах) от природы растворяющегося металла, в частности от величин его равновесного потенциала и анодной поляризации. В этом легко убедиться, если построить коррозионные диаграммы для трех различных металлов М, M1 и М2 (штрих-пунктирные линии на рисунке 5). На коррозию с кислородной деполяризацией может накладываться коррозия за счет выделения водорода, если равновесный потенциал водородного электрода в данном растворе положительнее равновесного потенциала корродирующего металла (прямые 1, 2 и 3 на рисунке 5). Величина предельного тока определяется растворимостью кислорода и величиной его коэффициента диффузии, но не зависит от природы металла, на котором восстанавливается кислород. В результате этого скорость процесса коррозии с кислородной деполяризацией меньше зависит от степени чистоты металла, чем скорость процесса коррозии с водородной деполяризацией, и изменяется в более широких пределах при изменении условий размешивания раствора и способа подвода кислорода (1,3-5).
6 Методы защиты металлов от коррозии
В зависимости от характера коррозии и условий ее протекания применяются различные методы защиты. Выбор того или иного способа определяется его эффективностью в данном конкретном случае, а также экономической целесообразностью. Любой метод защиты изменяет ход коррозийного процесса, уменьшая скорость или прекращая его полностью. Поляризационные или коррозионные диаграммы, наиболее полно характеризующие коррозионный процесс, должны отражать и те изменения в ходе его протекания, какие наблюдаются в условиях защиты. Поляризационные диаграммы можно использовать поэтому при разработке возможных путей предохранения металлов от коррозии. Они служат основой при выяснении принципиальных особенностей того или иного метода. В связи с этим при рассмотрении существующих методов защиты будем пользоваться поляризационными диаграммами в их несколько упрощенном виде (рисунок 6). На таких диаграммах постулируется линейная зависимость между плотностью тока и потенциалом каждой частной реакции. Это упрощение оказывается вполне допустимым при качественной оценке особенностей большинства методов защиты.
Эффективность защиты выражают при помощи коэффициента торможения у или степени защиты Z.
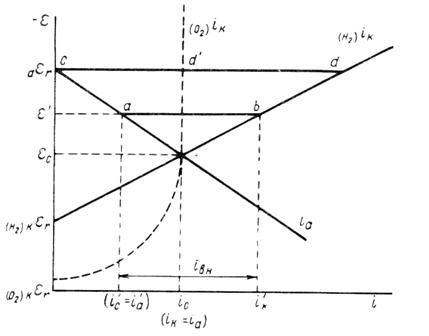
Рисунок 6 - Упрощенная поляризационная диаграмма коррозионного процесса, протекающего с водородной деполяризацией
Коэффициент торможения показывает, во сколько раз уменьшается скорость коррозии в результате применения данного способа защиты (уравнение 16).
ϒ = ic/i׳c(16)
где ic и i׳c — скорость коррозии до и после защиты. Степень защиты указывает, насколько полно удалось подавить коррозию благодаря применению этого метода (уравнения 17 и 18).
Z = (ic - i׳c)/ic(17)
или
Z% = (ic - i׳c) 100/ic(18)
Все методы защиты условно делятся на следующие группы:
- электрические методы;
- методы, связанные с изменением свойств корродирующего металла;
- методы, связанные с изменением свойств коррозионной среды;
- комбинированные методы.
Электрические методы защиты основаны на изменении электрохимических свойств металла под действием поляризующего тока. Наибольшее распространение получила защита металлов при наложении на них катодной поляризации. При смещении потенциала металла в сторону более электроотрицательных значений (по сравнению с величиной стационарного потенциала коррозии) скорость катодной реакции увеличивается, а скорость анодной падает (рисунок 6). Если при εс соблюдалось равенство
ik =ia, то при более отрицательном значении ε' это равенство нарушается -
i׳k ≠i׳a, причем i׳k>i׳a.
Уменьшение скорости анодной реакции при катодной поляризации эквивалентно уменьшению скорости коррозии. Коэффициент торможения при выбранном потенциале ε' (рисунок 4) будет
ϒ = ic/i׳c = ic/i׳а =ic/0,5iс= 2,
а степень защиты достигает 50%:
Z = (ic - i׳c) 100/ic = (ic – i׳а) 100/ic = (ic – 0,5ic) 100/ic =50%
Внешний ток iвн , необходимый для смещения потенциала до значения ε', представляет собой разницу между катодным и анодным токами:
iвн = i׳к – i׳а;
его величина на рисунке 6 выражается прямой ab. По мере увеличения внешнего тока потенциал смещается в более отрицательную сторону и скорость коррозии должна непрерывно падать. Когда потенциал корродирующего металла достигает равновесного потенциала анодного процесса аεг, скорость коррозии сделается равной нулю (ic = iа = 0), коэффициент торможения - бесконечности, а степень защиты - 100%. Плотность тока, обеспечивающая полную катодную защиту, называется защитным током iз. Его величине на рисунке 6 соответствует отрезок cd. Величина защитного тока не зависит от особенностей протекания данной анодной реакции, в частности от величины сопровождающей ее поляризации, а целиком определяется катодной поляризационной кривой. Так, например, при переходе от водородной к кислородной деполяризации сила защитного тока уменьшается и становится равной предельному диффузионному току (отрезок cd' на рисунке 6).
Защита металла катодной поляризацией применяется для повышения стойкости металлических сооружений в условиях подзем