


Современные методы фармацевтического анализа
СОВРЕМЕННЫЕ МЕТОДЫ ФАРМАЦЕВТИЧЕСКОГО АНАЛИЗА
1 Специфические особенности фармацевтического анализа
Фармацевтический анализ — это наука о химической характеристике и измерении биологически активных веществ на всех этапах производства: от контроля сырья до оценки качества полученного ЛВ, изучения его стабильности, установления сроков годности и стандартизации ЛФ. Фармацевтический анализ имеет свои специфические особенности, отличающие его от других видов анализа. Эти особенности заключаются в том, что анализу подвергают вещества различной химической природы: неорганические, элементорганические, радиоактивные, органические соединения от простых алифатических до сложных природных биологически активных веществ. Чрезвычайно широк диапазон концентраций анализируемых веществ. Объектами фармацевтического анализа являются не только индивидуальные ЛВ (субстанции), но и смеси, содержащие различное число компонентов.
Способы фармацевтического анализа нуждаются в систематическом совершенствовании в связи с созданием новых Л С и непрерывным повышением требований к их качеству. Причем растут требования как к степени чистоты ЛВ, так и к количественному содержанию. Поэтому необходимо широкое использование для оценки качества ЛС не только химических, но и более чувствительных физико-химических методов.
К фармацевтическому анализу предъявляют высокие требования. Он должен быть достаточно специфичен и чувствителен, точен по отношению к нормативам, обусловленным ГФ, ФС и другой НД, выполняться в короткие промежутки времени с использованием минимальных количеств испытуемых Л С и реактивов.
Фармацевтический анализ в зависимости от поставленных задач включает различные формы контроля качества ЛС: фармакопейный анализ, постадийный контроль производства ЛВ, анализ ЛФ индивидуального изготовления, экспресс- анализ в условиях аптеки и биофармацевтический анализ.
Составной частью фармацевтического анализа является фармакопейный анализ. Он представляет собой совокупность способов исследования ЛВ и ЛФ, изложенных в Государственной фармакопее или другой нормативной документации. На основании результатов, полученных при выполнении фармакопейного анализа, делается заключение о соответствии Л С требованиям ГФ (ФС, ФСП). При отклонении от этих требований ЛС к применению не допускают.
Заключение о качестве ЛС можно сделать только на основании анализа пробы (выборки). Порядок ее отбора указан либо в частной ФС, либо в общей статье ГФ XI (вып. 2).
Выполнение фармакопейного анализа позволяет установить подлинность Л В, его чистоту, определить количественное содержание фармакологически активного вещества или ингредиентов, входящих в состав Л Ф. Несмотря на то что каждый из этих этапов имеет свою конкретную цель, их нельзя рассматривать изолированно. Они взаимосвязаны, взаимно дополняют друг друга и отражают комплексный характер оценки качества ЛС. Так, например, температура плавления, растворимость, рН среды водного раствора и т.д. являются критериями как подлинности, так и чистоты ЛВ. Указанные особенности фармакопейного анализа существенно отличают его от норм и требований к методам анализа, используемых в Государственных стандартах (ГОСТ) и технических условиях (ТУ).
В ФС описаны методики соответствующих испытаний применительно к тому или иному фармакопейному ЛС. Многие из этих методик идентичны. В целях унификации способов анализа в ГФ включены общие фармакопейные статьи (ОФС), в которых систематизированы сведения о выполнении испытаний на ряд ионов и функциональных групп, а также единых методов количественного определения. Для обобщения большого объема частных сведений по фармакопейному анализу будут рассмотрены основные критерии фармацевтического анализа и общие принципы испытаний на подлинность, чистоту и количественного определения ЛВ.
2 Критерии фармацевтического анализа
На различных этапах фармацевтического анализа в зависимости от поставленных задач имеют значение такие критерии, как избирательность, чувствительность, точность, время, затраченное на выполнение анализа, израсходованное количество анализируемого ЛВ или ЛФ.
Избирательность метода очень важна при проведении анализа смесей веществ, поскольку дает возможность получать истинные значения каждого из компонентов. Только избирательные методики анализа позволяют определять содержание основного компонента в присутствии продуктов разложения и других примесей.
Требования к точности и чувствительности фармацевтического анализа зависят от объекта и цели исследования. При испытании степени чистоты ЛВ используют методики, отличающиеся высокой чувствительностью, позволяющие устанавливать минимальное содержание примесей.
При выполнении постадийного контроля производства, а также при проведении экспресс-анализа в условиях аптеки важную роль имеет фактор времени, которое затрачивается на выполнение анализа. Для этого выбирают методы, позволяющие провести анализ в наиболее короткие промежутки времени и вместе с тем с достаточной точностью.
При количественном определении ЛВ используют метод, отличающийся избирательностью и высокой точностью. Чувствительностью метода пренебрегают, учитывая возможность выполнения анализа с большой навеской ЛВ.
Мерой чувствительности реакции является предел обнаружения. Он означает наименьшее содержание, при котором поданной методике можно обнаружить присутствие определяемого компонента с заданной доверительной вероятностью. Термин «предел обнаружения» введен вместо такого понятия, как «открываемый минимум», им пользуются также взамен термина «чувствительность». На чувствительность качественных реакций оказывают влияние такие факторы, как объемы растворов реагирующих компонентов, концентрации реактивов, рН среды, температура, продолжительность опыта. Для установления чувствительности реакций все шире используют показатель поглощения (удельный или молярный), устанавливаемый спектрофотометрическим методом. Высокой чувствительностью отличаются физико-химические методы анализа. Наиболее высокочувствительны радиохимические и масс-спектральный методы, позволяющие определять 10~8-10~9 г анализируемого вещества, полярографические и флуориметрические (10~6-10"9 г); чувствительность спек- трофотомстрических методов Ю-3-10 6, потенциометрических — Ю"2 г.
Термин «точность анализа» включает одновременно два понятия: воспроизводимость и правильность полученных результатов. Воспроизводимость характеризует рассеивание результатов анализа по сравнению со средним значением. Правильность отражает разность между действительным и найденным содержанием вещества. Точность анализа у каждого метода различна и зависит от многих факторов: калибровки измерительных приборов, точности отвешивания или отмеривания, опытности аналитика и т.д. Точность результата анализа не может быть выше, чем точность наименее точного измерения.
Так, при вычислении результатов титриметрических определений наименее точная цифра — количество миллилитров ти- транта, израсходованного на титрование. В современных бюретках в зависимости от класса их точности максимальная ошибка отмеривания около ±0,02 мл. Ошибка от натекания тоже равна ±0,02 мл. Если при указанной общей ошибке отмеривания и натекания ±0,04 мл на титрование расходуется 20 мл тшпранта, то относительная погрешность составит 0,2%. Приуменьшении навески и количества миллилитров титранта точность соответственно уменьшается. Таким образом, ти- триметрическое определение можно выполнять с относительной погрешностью +(0,2-0,3)%.
Точность титриметрических определений можно повысить, если пользоваться микробюретками, применение которых значительно уменьшает ошибки от неточного отмеривания, натекания и влияния температуры. Погрешность допускается также при взятии навески.
Отвешивание навески при выполнении анализа ЛВ осуществляют с точностью до +0,2 мг. При взятии обычной для фармакопейного анализа навески 0,5 г ЛВ и точности взвешивания ±0,2 мг относительная погрешность будет равна 0,4%. При анализе ЛФ и выполнении экспресс-анализа такая точность при отвешивании не требуется, поэтому навеску берут с точностью +(0,001-0,01) г, т.е. с предельной относительной погрешностью 0,1-1%.
При выполнении количественного определения любым химическим или физико-химическим методом могут быть допущены три группы ошибок: грубые (промахи), систематические (определенные) и случайные (неопределенные).
Грубые ошибки являются результатом просчета наблюдателя при выполнении какой-либо из операций определения или неправильно выполненных расчетов. Результаты с грубыми ошибками отбрасываются как недоброкачественные.
Систематические ошибки отражают правильность результатов анализа. Они искажают результаты измерений обычно в одну сторону (положительную или отрицательную) на некоторое постоянное значение. Причиной систематических ошибок в анализе могут быть, например, гигроскопичность ЛВ при отвешивании его навески, несовершенство измерительных и физико-химических приборов, опытность аналитика и т.д. Систематические ошибки можно частично устранить внесением поправок, калибровкой прибора и т.д.
Случайные ошибки отражают воспроизводимость результатов анализа. Они вызываются неконтролируемыми переменными.
Правильность результатов определений выражают абсолютной ошибкой и относительной ошибкой (погрешностью).
Абсолютная ошибка представляет собой разность между полученным результатом и истинным значением. Эта ошибка выражается в тех же единицах, что и определяемая величина (граммах, миллилитрах, процентах).
Относительная погрешность определения равна отношению абсолютной ошибки к истинному значению определяемой величины. Выражают относительную погрешность обычно в процентах (умножая полученную величину на 100). Относительная погрешность определений физико-химическими методами включает как точность выполнения подготовительных операций (взвешивание, отмеривание, растворение), так и точность выполнения измерений на приборе (инструментальная ошибка).
Индивидуальные вещества можно определять при анализе спектрофотометрическим методом в УФ- и видимой областях с относительной погрешностью ±(2-3)%, ИК-спектрофотометрией ±(5-12)%, газожидкостной хроматографией = (3-3,5)%, полярографией ±(2-3)%, потенциометрией ±(0,3-1)%. При анализе ЛВ в многокомпонентных смесях относительная погрешность определения этими методами возрастает примерно в два раза. Сочетание хроматографии с другими методами позволяет выполнять анализ Л В в ЛФ с относительной погрешностью ±(3-7)%.
Точность биологических методов намного ниже, чем химических и физико-химических. Относительная погрешность биологических определений достигает 20-30 и даже 50%. Для повышения точности в ГФ XI введен статистический анализ результатов биологических испытаний.
Относительная погрешность определения может быть уменьшена за счет увеличения числа параллельных измерений. Однако эти возможности имеют определенный предел. Уменьшать случайную ошибку измерений, увеличивая число опытов, целесообразно до тех пор, пока она станет меньше систематической. Обычно в фармацевтическом анализе для расчетов берут среднее из трех параллельных измерений. При статистической обработке результатов определений с целью получения достоверных результатов выполняют не менее семи параллельных измерений.
3 Физические свойства, используемые для установления подлинности лекарственных веществ
Испытание на подлинность — это подтверждение идентичности анализируемого лекарственного вещества (лекарственной формы), осуществляемое на основе требований Фармакопеи или другой НД (ФС, ФСП). Испытания выполняют физическими, химическими и физико-химическими методами. Свои особенности имеют испытания неорганических, элементорганических и органических ЛВ.
Подлинность ЛВ подтверждают показатели: описание внешнего вида, его физические свойства, физические константы и растворимость в различных растворителях. Они дают ориентировочную характеристику испытуемого ЛВ.
Физические свойства твердых ЛВ оценивают по форме кристаллов или по виду аморфного вещества, его устойчивости к свету, кислороду, содержащемуся в воздухе, гигроскопичности и степени выветривания, запаху, цвету, степени белизны. Степень белизны (по ГФ XI) Л В оценивают на спектрофотометрах СФ-18 по спектру отражения образца при его освещении белым светом. Для жидкостей устанавливают цвет, запах, летучесть, подвижность, воспламеняемость.
Температура плавления — это температура, при которой вещество переходит из твердого состояния в жидкое. По ГФ XI под температурой плавления понимают интервал температур между началом плавления (появление первой капли жидкости) и концом плавления (полным переходом вещества в жидкое состояние). Интервал между началом и концом плавления не должен превышать 2°С. Температура плавления — постоянная характеристика для индивидуального ЛВ. В присутствии даже небольшого количества примесей она изменяется, что используется для подтверждения степени чистоты Л В.
Для ЛВ, неустойчивых при нагревании, согласно требованиям ГФ XI устанавливают температуру разложения, т.е. температуру, при которой происходит резкое изменение вещества (вспенивание). Если переход вещества из твердого в жидкое состояние нечеткий, то устанавливают только температуру начала или температуру конца плавления, что оговаривается в ФС или ФСП.
В ГФ XI (вып. I, с. 17) приведены три метода определения температуры плавления. Применение того или иного метода зависит от физических свойств веществ: метод 1 и 1а применяют для легкорастираемых в порошок твердых Л В, устойчивых (метод 1) и неустойчивых (метод 1а) при нагревании; методы 2 и 3 используют для Л В, не растирающихся в порошок (жпры, воск, парафин, вазелин, смолы).
Температура затвердевания — наиболее высокая температура, при которой в течение короткого времени происходит переход ЛВ из жидкого в твердое состояние.
Температуру кипения устанавливают для жидких ЛВ. Это температура, при которой жидкость превращается в пар. Для практических целей по ГФ XI используют температурные пределы перегонки — интервал между начальной и конечной температурой кипения при нормальном атмосферном давлении 101,3 кПа (760 мм рт. ст.). Начальной считают температуру кипения, при которой в приемник перегоняются первые 5 капель жидкости, а конечной — 95% жидкости.
Плотностью называют массу единицы объема вещества (массу I см3) при стандартной температуре (обычно 20°С). Определение плотности проводят с помощью пикнометра в тех случаях, когда следует установить эту константу с точностью до 0,001, или ареометра (в случае определения плотности с точностью до 0,01). Соответствующие методики описаны в ГФ XI (в. 1, с. 24-26).
Вязкость (внутреннее трение) — свойство текучих тел (жидкостей) оказывать сопротивление перемещению одной их части относительно другой при определенной температуре. Для подтверждения качества жидких ЛВ, имеющих вязкую консистенцию, обычно определяют относительную вязкость (По™.), принимая вязкость воды за единицу. Различают также динамическую (абсолютную), удельную, приведенную, характеристическую и кинематическую вязкость. Последнюю устанавливают с помощью вискозиметра Оствальда (ГФ XI, в. 1, с. 87-94).
Растворимость — свойство газообразных, жидких и твердых веществ переходить в растворенное состояние. Растворимость в фармакопейном анализе рассматривают как свойство ЛВ растворяться в различных растворителях. Растворимость прп постоянной температуре является одной из основных характеристик, с помощью которой подтверждают доброкачественность большинства ЛВ.
Для обозначения растворимости в ГФ XI приняты условные термины, указывающие количество растворителя (мл), необходимое для растворения 1 г ЛВ. Различают очень легко растворимые (до 1 мл), легко растворимые (от 1 до 10), растворимые (от 10 до 30), умеренно растворимые (от 30 до 100), мало растворимые (от 100 до 1000), очень мало растворимые (от 1000 до 10 000), практически нерастворимые (более 10 000 мл).
Методика определения растворимости по ГФ XI (вып. 1, с. 175-176) состоит в том, что навеска ЛВ вносится в отмеренный объем растворителя и непрерывно перемешивается в случае необходимости до 10 мин. при 20 ± 2°С. Растворившимся Л В считают в том случае, если в растворе при наблюдении в проходящем свете не наблюдается частиц вещества. Отклонения от этого общего правила: образование мутных растворов, растворение более продолжительное, чем в течение 10 мин. 'такие ЛВ называют медленно растворимыми). Показатели растворимости в различных растворителях указываются в ФС. В качестве растворителей, кроме воды, используются растворы кислот и щелочей (карбонатов), а также различные органические растворители (этанол, метанол, хлороформ, эфир, ацетон, гексан, дихлорэтан, этилацетат) и масла.
Метод фазовой растворимости основан на правиле фаз Гиббса, которое устанавливает зависимость между числом фаз и числом компонентов в условиях равновесия. Суть установления фазовой растворимости состоит в последовательном прибавлении увеличивающейся массы ЛВ к постоянному объему растворителя при постоянной температуре и непрерывном встряхивании. Затем с помощью диаграмм количественно определяют массу растворенного ЛВ, устанавливая процентное содержание в нем примеси. Таким образом, метод фазовой растворимости позволяет осуществить количественную оценку степени чистоты ЛВ путем точных измерений значений растворимости. Он применим ко всем соединениям, которые образуют истинные растворы, и используется для изучения стабильности и получения очищенных (до 99,5%) образцов ЛВ.
4 Химические методы установления подлинности
Идентификация неорганических лекарственных веществ
Установление подлинности неорганических Л В основано на обнаружении с помощью химических реакций катионов и анионов, входящих в состав их молекул. С точки зрения приемов выполнения испытаний и получаемых при этом результатов можно выделить несколько общих способов.
Реакции осаждения основаны на образовании нерастворимых в воде продуктов реакции, аналитический эффект можно охарактеризовать по окраске или по растворимости осадков (в органических растворителях, кислотах, щелочах).
Эту же реакцию используют для обнаружения фосфат-ионов. Арсенат-иопы дают аналогичные результаты:
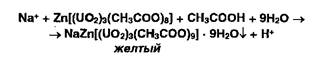
Попы калия осаждают винной кислотой:
![]()
Ионы натрия осаждают цинкуранилацетатом:
![]()
Ионы калия мож-чо осадить раствором гексанитрокобальтата (III) натрия:
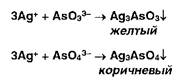
Некоторые реакции осаждения можно использовать для обнаружения и катионов, и анионов.
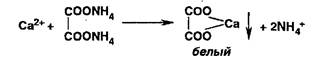
Попы магния образуют в присутствии фосфат- и аммоний-ионов осадок фосфата магния-аммония:
![]()
![]()
Фосфат-ионы образуют с раствором молибдата аммония желтый осадок фосфор-молибдата аммония:
![]()
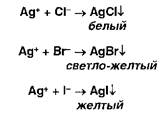
![]()
![]()
![]()
![]()
![]()
Ионы бария образуют белый осадок с сульфат-ионами: Аналогичную реакцию дают сульфиты:
Сульфит бария, в отличие от сульфата бария, растворим в хлороводородной кислоте. Ионы фтора открывают реакцией осаждения из раствора хлорида кальция:
Ионы серебра образуют осадки с хлоридами, бромидами, йодидами:
Образующиеся галогениды различаются по растворимости в растворе аммиака. Желтый осадок образуют ионы серебра с фосфатами:
Образует осадки ион серебра также с арсенит- и арсенат-ионами:
Ионы магния с растворами карбонатов образуют белый осадок основного карбоната магния:
Ионы железа (III) в растворе приобретают красное окрашивание в присутствии роданид-ионов, образуя малодис- социирующее соединение:
![]()
Ряд реактивов образуют белые или окрашенные осадки с несколькими катионами. Ионы ртути, цинка, висмута, мышьяка взаимодействуют с сульфидами:

Ионы железа (III) и цинка осаждаются растворами гсксацианоферрата (II) калия:
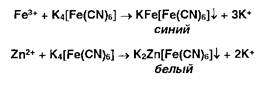
Ионы железа (II) дают аналогичные результаты с гексацианоферратом (III) калия:
![]()
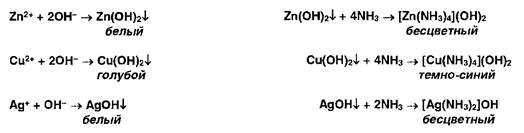
Ионы цинка, меди и серебра осаждаются гидроксидом аммония с образованием осадков, растворимых в избытке г г-ктива:
Ионы ртути (II) и висмута (III) осаждаются йодидами, осадки растворяются в избытке реактивов:

Окислительно -восстановительные реакции, используемые для испытаний подлинности, сопровождаются изменением окраски образующихся продуктов взаимодействия.
Бромид- и йодид-ионы окисляют хлором (хлорамином, другими окислителями):
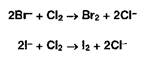
Выделившийся бром окрашивает слой хлороформа в оранжевый цвет, а йод — в фиолетовый. Йод обнаруживают также по синему окрашиванию крахмального клейстера.
Фторид-ионы обесцвечивают красную окраску раствора роданида железа: ![]() Ионы меди, серебра восстанавливаются из оксидов и солей до свободных металлов:
Ионы меди, серебра восстанавливаются из оксидов и солей до свободных металлов:
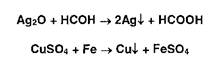
Нитрат- и нитрит-ионы обнаруживают путем окисления дифениламина до дифенилбензидина, а затем до дифе- нилдифенохинондиимина гидросульфата (синее окрашивание) в присутствии концентрированной серной кислоты:
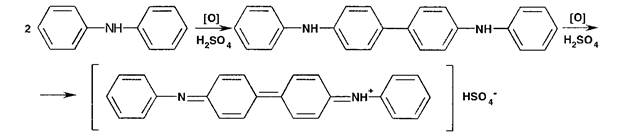
Нитрит-иоиы (в отличие от нитратов) обесцвечивают раствор перманганата калия, подкисленный серной кислотой:
![]()
![]()
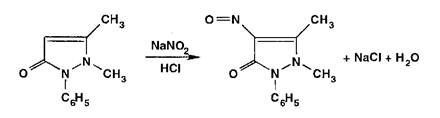
Взаимодействуя с антипирином (феназоном), нитриты образуют продукт замещения — нитрозоантипирин (зеленое окрашивание):
Реакции разложения сопровождаются образованием газообразных продуктов, которые обнаруживают органолеп- тически (запах, окраска).
Ионы аммония разлагаются под действием растворов гидроксидов (запах аммиака или изменение окраски красной лакмусовой бумаги):
Карбонат-ионы под действием насыщенного раствора сульфата магния образуют белый осадок, а гидрокарбонат образует осадок только при кипячении смеси (см. реакцию на магний).
Карбонат- и гидрокарбонат-ионы образуют газ — диоксид углерода под действием минеральных кислот:
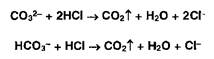
Сульфит-ионы в тех же условиях образуют диоксид серы (резкий запах):
![]()
Нитрит-ионы, в отличие от нитрат-ионов, под действием кислот выделяют оксиды азота (диоксид азота имеет красно-бурую окраску):
![]()
Превращения, происходящие при нагревании и прокаливании некоторых ЛВ. Йод кристаллический, соединения мышьяка, ртути возгоняются (испытания выполнять под тягой!). Цинка оксид при прокаливании желтеет (после охлаждения окраска исчезает). Висмута нитрат основной разлагается с образованием оксида висмута (желтое окрашивание) и диоксида азота (желто-бурые пары). Соли алюминия при прокаливании с нитратом кобальта образуют плав синего цвета, представляющий собой алюминат кобальта (тенарова синь). Соли цинка в этих условиях образуют плав зеленого цвета (зелень Ринмана).
Установить наличие ряда элементов в неорганических и элементорганических ЛВ можно по изменению окраски бесцветного пламени горелки. Так, соль натрия, внесенная в пламя, окрашивает его в желтый цвет, калия — в фиолетовый, кальция — в кирпично-красный, лития — в карминово-красный. Соли бора, смоченные этанолом, окрашивают кайму пламени в зеленый цвет.
5 Идентификация элементорганических лекарственных веществ
Поскольку атомы у большинства элементорганических соединений связаны ковалентно, необходимым условием испытания их подлинности является предварительная минерализация. При этом происходит частичное или полное разрушение органической части молекулы до оксида углерода (IV) и воды. Другие элементы образуют соответствующие ионы. Последние идентифицируют с помощью рассмотренных выше или иных реакций.
Серу обнаруживают либо путем восстановления до сульфид-ионов, либо окислением до сульфат-ионов. Образование сульфида происходит также из соединений, содержащих тиоэфирную или тиокетонную серу, при нагревании с 10% раствором гидроксида натрия:
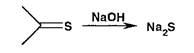
Образовавшийся при восстановлении органически связанной серы сульфид натрия идентифицируют цветной реакци- с% с н игропруссидом натрия (красно-фиолетовое окрашивание), осаждением раствором соли свинца (черное) или по сероводорода:
![]()
Счисление органически связанной серы осуществляют действием концентрированной азотной кислоты или сплавле- - .-ем со смесью нитрата и карбоната калия. Образовавшийся сульфат-ион открывают реакцией с солями бария.
Фосфорсодержащие соединения минерализуют смесью концентрированных серной и азотной кислот до фосфат-ионов, чсторые обнаруживают реакциями образования фосфата магния-аммония или фосфор-молибдата аммония (см. реакции на фосфат-ион).
Галогенсодержащие соединения под действием цинковой пыли в кислой или щелочной среде образуют галогениды:
![]()
Затем обнаруживают образовавшиеся галогенид-ионы с помощью рассмотренных выше реакций. Проба Бейльштейна основана на образовании окрашенных в зеленый цвет галогенидов меди при внесении в бесцветное пламя медной проволоки с галогенсодержащим соединением.
Фтор н хлор открывают аналитическими реакциями на соответствующие ионы после разрушения органической части молекулы расплавленным металлическим натрием:
![]()
Йод обнаруживают либо нагреванием йодпроизводного в пробирке на пламени горелки, либо действуя концентрированной серной кислотой:
![]()
Наблюдают выделение фиолетовых паров йода или фиолетовую окраску хлороформного извлечения. Можно также применить спекание со смесью нитрата калия и карбоната натрия:
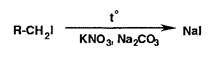
Затем обнаруживают йодид-ионы.
Метод спекания можно использовать при наличии в одном соединении хлора и серы с последующим обнаружением образовавшихся хлорид- и сульфат-ионов.
Кобальт обнаруживают в виде ионов реакцией с нитрозо-Я-солью (динатриевой солью 1-нитрозо-2-нафтол-3,6-ди- . "ьфокислоты) после спекания кобальтсодержащего соединения с гидросульфитом калия (красное окрашивание).
6 Идентификация органических лекарственных веществ
В ~армацевтическом анализе используются различные химические реакции органических соединений, которые дают определенный аналитический эффект (выпадение осадка, выделение газа, образование окрашенного раствора и т.д.).
Реаыши нитрования сопровождаются образованием окрашенных в желтый цвет моно-, ди- и тринитропроизводных арома-",'- еекого ряда:
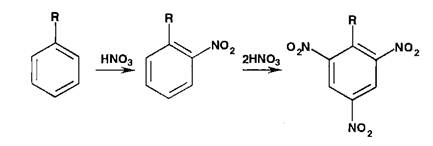
Под действием гидроксидов калия (натрия) продукты нитрования образуют окрашенные ацисоли:
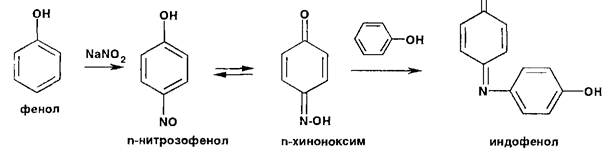
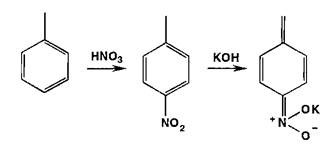
Реакции нитрозироваиия приводят к образованию окрашенных, флюоресцирующих или имеющих стабильную температуру плавления нитрозосоединений:
Фенолы образуют нитрозосоединения, бесцветные или окрашенные в сине-зеленый (фенол), сине-фиолетовый (резорцин) цвет. При нитрозировании фенолов с последующим окислением образуются индофенолы (интенсивно-синее окрашивание):
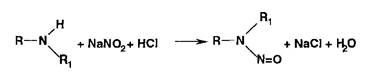
Реакции диазотирования и азосочетания используют для идентификации производных первичных ароматических аминов и фенолов. Азосоединения — окрашенные (в красный, коричневый и оранжевый цвет) продукты, получаемые в две стадии:
1. Диазотирование (получение соли диазония):
![]()
2. Азосочетание (взаимодействие соли диазония с фенолом или ароматическим амином). Сочетание происходит в ор- пю- или паря-положеннях по отношению к гидроксильной или аминогруппе, но идет легче в псрс-положении:
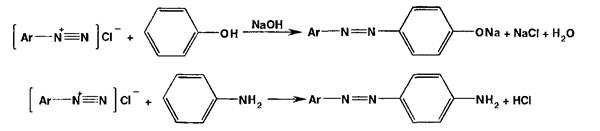
Аз-зс;че_гч".'.е с фенолами (нафтолами) происходит в слабощелочной (рН 9,0-10,0), а с аминами — в слабокислой сре-
Т.?: 1;: сочетания обусловлен наличием в этих соединениях электронодонорных -ОН и -ИН2 групп, создающих : -?.■■..отельные заряды в орто- и пара-положениях ароматического ядра. В этих положениях происходит электро- г ~ъг : е :гч?_;гние водорода катионом диазония и образуется азосоединение.
Г -: лзссочетания используют также для идентификации сложных эфиров фенолов, ацилированных первичных сг : • аминов (после гидролиза) и нитропроизводных (после гидрирования).
Раилии галогенирования (бромирования и йодирования) по типу реакции элеюрофильного замещения используют для обпроизводных фенолов и первичных ароматических аминов. Наличие в их молекулах заместителей первого рода (оксл- и аминогруппы) обусловливает происходящий процесс образования трибромфенола или триброманилина (белый осадок):
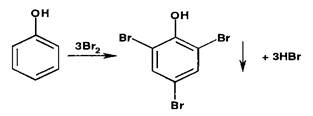
Аналогично происходит процесс образования трийодпроизводных. При наличии в молекулах фенола и анилина радикалов в пара- или орто-положениях образуются моно- или дигалогенпроизводные.
Реакции дегалогенирования можно выполнять без предварительной минерализации (если галогены связаны с углеродом непрочной ковалентной связью). Отщепление галогена при этом происходит под действием раствора нитрата серебра:
![]()
Дегалогенируют также, используя щелочное отщепление, путем нагревания галогенпроизводного в присутствии цинковой пыли (бромкамфора) или в спиртовом растворе гидроксида натрия:
![]()
Затем обнаруживают галогенид-ион.
Реакции конденсации альдегидов и кетонов с первичными аминами, гидроксиламином, гидразинами используются для идентификации всех указанных групп органических соединений по общей схеме:
![]()
Альдегиды, конденсируясь с первичными аминами, образуют окрашенные в желтый, красный или оранжевый цвет соли оснований Шиффа:
![]()
Эта реакция лежит в основе лигниновой пробы на первичные ароматические амины, которые взаимодействуют с ли- гнинами, содержащимися в бумаге.
Кетопроизводные образуют гидразоны:
![]()
и кетоксимы:
![]()
Гидразоны и кетоксимы — белые или окрашенные нерастворимые в воде соединения со стабильной температурой плавления. По этим признакам можно идентифицировать исходные для их получения соединения.
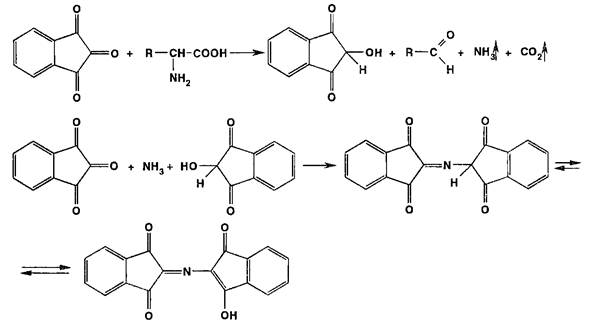
Окислительная конденсация с участием альдегидов лежит в основе таких широко применяемых в фармацевтическом анализе реакций, как образование ауринового красителя, нингидриновая реакция, мурексидная проба, проба Ле Розена и др.
Нингидриновая реакция является общей для а-аминокислот, иминокислот, полипептидов. Нингидрин (1,2,3-трикето- гидринденгидрат) образует с аммиаком, выделившимся из этих соединений, продукт конденсации — ион дикетогидрин- дилидендикетогидрамина, имеющий сине-фиолетовое окрашивание:
Реакции этерификации, ацилирования и гидролиза. Для подтверждения подлинности спиртов и карбоновых кислот широко используют реакцию этерификации, а подлинность сложных эфиров подтверждают с помощью обратного процесса — гидролиза:
![]()
![]()
Этерификацию проводят в присутствии дегидратирующих веществ (концентрированная серная кислота), а гидролиз — в кислой или щелочной среде.
Сходен с этерификацией процесс ацилирования (особенно ацетилирования) аминопроизводных:
а также обратный процесс — гидролиз ацильных производных.
Образовавшиеся в результате этерификации, ацилирования, гидролиза продукты идентифицируют по аналитическому эффекту (цвету, запаху, образованию газа или осадка, температуре плавления осадка и др.).
Очень широко используют, например, реакцию образования этилацетата, имеющего своеобразный фруктовый запах. Этилацетат образуют органические соединения, выделяющие при гидролизе этанол или уксусную кислоту:
![]()
Общим способом испытаний ЛВ, содержащих в молекуле сложноэфирную, лактонную, лактамную, амидную, имид- ную группы, является реакция, основанная на образовании гидроксамовых кислот (гидроксамовая проба):
![]()
Гидроксамовые кислоты, взаимодействуя с ионами железа (III) или меди (II), образуют окрашенные соли:
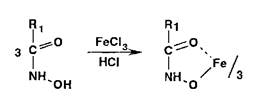
Реакции разложения амидов происходят при нагревании в растворах едких щелочей с образованием аммиака или алки- ламинов. имеющих характерный запах:
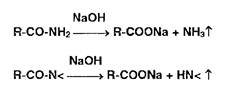
Первичные, вторичные и третичные амины в тех же условиях образуют соответственно метиламин, диметиламин ••" гриметиламин, например:
![]()
![]()
Указанные химические реакции используют для испытания подлинности солей первичных аммониевых оснований, амидов ароматических и гетероциклических кислот, производных уретанов.
Ациклические и циклические уреиды, алкилуреиды сульфокислот, производные гуанидина и семикарбазона, имеющие в молекуле уреидную группу, гидролизуются в щелочной среде с образованием аммиака. Например, уреиды:
К этой группе реакций можно отнести используемый в фармацевтическом анализе пиролиз (термическое разложение в сухой пробирке). Используют пиролиз для идентификации сульфаниламидов, производных бензодиазепина, пиридина и других ЛВ, которые образуют плавы с различной окраской и выделяют газообразные продукты с характерным запахом.
Реакции окисления-восстановления.
Процесс гидрирования осуществляют, как правило, водородом в момент выделения (при взаимодействии металлического цинка с хлороводородной кислотой). Эту реакцию используют для идентификации непредельных соединений, превращая их в предельные, или для восстановления нитросоединений до аминопроизводных:
![]()
Широко используются в фармацевтическом анализе реакции окисления. Первичные спирты идентифицируют, последовательно окисляя до альдегидов и кислот, которые затем обнаруживают с помощью характерных реакций:
![]()
Так, например, восстановительные свойства альдегидов устанавливают с помощью реакции образования «серебряного зеркала»:
![]()
Этот же процесс лежит в основе взаимодействия реактива Несслера с альдегидами:
![]()
Реакция окисления альдегидов лежит в основе использования реактива Фелинга, представляющего собой смесь от- лельно приготавливаемых растворов сульфата меди и калий-натриевой соли винной кислоты. В щелочной среде при нагревании в присутствии альдегидов образуется красный осадок оксида меди (I). Общая схема этой реакции:
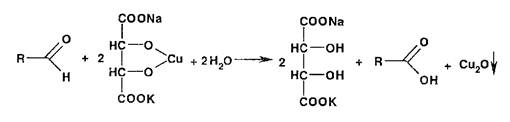
8 Реакции образования солей и комплексных соединений
Соли органических кислот идентифицируют по наличию катионов натрия, калия, кальция и др. (с помощью рассмотренных выше реакций), а также по наличию анионов органических кислот (ацетат-,