


Методы определения активности катализаторов
Федеральное агентство по образованию Российской Федерации
Казанский государственный технологический университет
Кафедра ТООНС
РЕФЕРАТ
по курсу: катализ в органической технологии на тему:
Методы определения активности катализаторов.
Казань 2007 г.
ВОЗНИКНОВЕНИЕ И РАЗВИТИЕ КАТАЛИЗА
Катализаторы играют исключительно важную роль в живой природе. Г.К. Боресков отмечает, что почти все реакции в живых организмах сами по себе происходят медленно и только благодаря участию биологических катализаторов протекают с достаточной для организмов скоростью. В состав многих катализаторов, стимулирующих природные процессы, входят металлы. Наиболее важными биологическими катализаторами для живых организмов являются ферменты (энзимы), которые с древних времен несознательно используются людьми при приготовлении вина и образовании уксуса из спиртовых жидкостей.(1)
Человечество может многому поучиться у живой природы в области катализа. Так, например, под действием катализаторов живой природы, азот воздуха вступает в реакцию при атмосферном давлении и обычной температуре, а ныне применяемые в промышленности катализаторы проявляют достаточную активность лишь при давлении в несколько сот атмосфер и температурах более 400°С. В то же время людям, создающим новые катализаторы, едва ли целесообразно полностью моделировать природные условия, поскольку они имеют возможность применять более высокие концентрации реагентов, температуры и давления, чем в обычных природных условиях.(1)
Катализ (от греч. katбlysis — разрушение), изменение скорости химических реакций в присутствии веществ (катализаторов), вступающих в промежуточное химическое взаимодействие с реагирующими веществами, но восстанавливающих после каждого цикла промежуточных взаимодействий свой химический состав. Реакции с участием катализаторов называются каталитическими. Количество реагирующего вещества, которое может испытать превращение в присутствии определённого количества катализатора, не ограничивается какими-либо стехиометрическими соотношениями и может быть очень большим. Этим каталитические реакции отличаются от индуцируемых, или сопряжённых реакций, когда одна реакция вызывается или ускоряется (индуцируется) другой и происходит необратимое превращение вещества-индуктора. Возможные изменения катализатора при каталитических реакциях являются результатом побочных процессов, ни в коей мере не обусловливающих каталитическое действие.(1)
Воздействие катализатора открывает новый реакционный путь, обычно с большим числом стадий, на котором катализатор входит в состав активного комплекса (активированного комплекса) по крайней мере одной из стадий. Если при этом скорость реакции становится больше, чем в отсутствие катализатора, то К. называется положительным (его нередко отождествляют с общим понятием К.). Возможен и обратный случай, когда происходит отрицательный К.: в присутствии катализатора исключается один из возможных путей реакции и остаются лишь более медленные, в результате чего реакция замедляется или даже практически полностью подавляется (см. Антиокислители, Ингибиторы химические). Особый случай К. — ускорение реакции при воздействии продукта реакции или одного из промежуточных веществ, образующихся при реакции (см. Автокатализ).(1)
К. не связан с изменением свободной энергии катализатора, и воздействие катализатора не может поэтому смещать положение равновесия химической реакции. Вблизи состояния равновесия катализаторы в равной степени ускоряют как прямую, так и обратную реакцию.(1)
Основным фактором, определяющим скорость химического превращения, является энергия активации (Е) — разность энергий активного комплекса и исходных реагирующих молекул. Если предположить, что реакция не нарушает равновесного распределения энергии между молекулами, то вероятность образования активного комплекса, а следовательно, и скорость реакции в первом приближении пропорциональна exp (—E/RT), где R — газовая постоянная, Т — абсолютная температура. Отсюда следует, что скорость реакции тем больше, чем меньше Е, и вследствие экспоненциальной зависимости возрастает значительно даже при небольшом снижении Е. На рис. представлено изменение энергии при реакции без катализатора (кривая 1) и при участии катализатора (кривые 2 и 3). Кривая 2 с двумя максимумами соответствует образованию одного промежуточного продукта. Число стадий и промежуточных продуктов часто бывает значительно большим. Взаимодействие реагирующих веществ с катализатором может и не приводить к образованию стабильной формы промежуточного соединения (кривая 3). Но и в этом случае катализатор входит в состав активного комплекса и взаимодействие реагирующих веществ с катализатором определяет реакционный путь. Если энергии активных комплексов всех стадий реакционного пути с участием катализатора ниже энергии активного комплекса реакции без катализатора (т. е., и E3 ниже E1), то участие катализатора приведёт к увеличению скорости (положительный К.). Во многих случаях К. ускорение реакции достигается благодаря появлению богатых энергией частиц в процессе самой реакции, причём их концентрация может превосходить равновесную (см. Цепные реакции). Например, каталитическое воздействие воды на окисление окиси углерода связано с развитием реакционных путей с участием гидроксильных групп и атомов водорода. Отрицательный К. часто связан с прекращением цепной реакции вследствие обрыва цепей при взаимодействии отрицательного катализатора с активными частицами. Примером может служить замедляющее влияние кислорода на соединение водорода с хлором.(1)
Характер промежуточного химического взаимодействия при К. весьма разнообразен. Обычно различают две группы каталитических процессов: кислотно-основной (гетеролитический) и окислительно-восстановительный (гомолитический). В процессах первой группы происходит промежуточное кислотно-основное взаимодействие реагирующих веществ с катализатором, например переход протона от катализатора к реагирующим веществам или наоборот. На последующих стадиях протон перемещается в обратном направлении, и катализатор восстанавливает свой состав. При К. апротонными кислотами взаимодействие осуществляется через свободную пару электронов реагирующего вещества. Примерами кислотно-основного К. могут служить гидролиз сложных эфиров, ускоряемый кислотами; гидратация олефинов в присутствии фосфорно-кислотных катализаторов; изомеризация и крекинг углеводородов на алюмосиликатных катализаторах; алкилирование; полимеризация и многие другие реакции. При реакциях окислительно-восстановительного К. промежуточное взаимодействие связано с электронными переходами между катализатором и реагирующими веществами. К этой группе относятся окисление двуокиси серы в трёхокись в производстве серной кислоты; окисление аммиака до окиси азота при получении азотной кислоты; многочисленные процессы парциального окисления органических соединений, например этилена в окись этилена, нафталина во фталевый ангидрид; гидрогенизация; дегидрогенизация; циклизация и ароматизация углеводородов; разложение перекиси водорода и многие др. Каталитической активностью в отношении окислительно-восстановительных реакций обладают преимущественно металлы 4-, 5- и 6-го периодов системы Д. И. Менделеева, имеющие недостроенную d-оболочку электронов, их соединения и в меньшей мере соединения элементов с достраивающейся f-оболочкой (лантаноиды и актиноиды).(1)
Рассмотренные группы далеко не охватывают всё разнообразие каталитических реакций. Характер промежуточного взаимодействия при К. гораздо более сложен и зависит от всех деталей электронной структуры как реагирующих веществ, так и катализатора. Конкретные механизмы каталитических реакций многообразны и пока лишь в немногих случаях твёрдо установлены.(1)
В зависимости от фазового состояния реагирующих веществ и катализатора различают гомогенный и гетерогенный К. Промежуточное положение занимает микрогетерогенный К. в коллоидных системах (например, К. ферментами). При гомогенном К. катализатор и реагирующие вещества образуют одну однородную систему, границы раздела между катализатором и реагирующими веществами отсутствуют. При гетерогенном К. катализатор и реагирующие вещества находятся в разных фазах и отделены друг от друга границей раздела. Наиболее важны случаи, когда катализатор является твёрдым телом, а реакционная система образует жидкую или газообразную фазу. Промежуточное взаимодействие происходит при этом преимущественно на поверхности твёрдого катализатора.(1)
Выбор состава катализатора для определённой реакции является очень сложной проблемой, решаемой пока главным образом эмпирическим путём. В СССР предложен и развит ряд теоретических подходов, основанных на корреляции отдельных частных свойств катализаторов с их активностью. Так, мультиплетная теория К. (первые публикации 1929) предполагает промежуточное взаимодействие реагирующих веществ с несколькими атомами на поверхности твёрдых катализаторов и придаёт решающее значение соответствию расстояний между атомами в молекулах реактантов и параметров кристаллической структуры катализатора. В дальнейшем теория была дополнена представлением о необходимости определённого соответствия энергий связей, разрывающихся и образующихся в результате реакции, и энергий связей реактантов с катализатором при промежуточном взаимодействии. Значительное распространение в 50-х гг. получило представление о зависимости каталитической активности твёрдых катализаторов, обладающих полупроводниковыми свойствами, от их электрических характеристик, — так называемая электронная теория К. По этой теории предполагается, что промежуточное взаимодействие реактантов с катализатором осуществляется при участии электронов проводимости твёрдого катализатора и поэтому зависит от его коллективных электронных свойств — расположения энергетических зон и локальных уровней электронов, работы выхода электрона, концентрации носителей тока и др. В гетерогенном К. широко использовалось предположение (выдвинутое в 1939) о существовании на поверхности твёрдых катализаторов особых активных центров, представляющих собой ребра, углы или различные структурные нарушения (дислокации) нормальной кристаллической структуры. Предполагалось также, что при нанесении каталитически активного вещества на инертный носитель особые каталитические свойства проявляют отдельно расположенные атомы или совокупности небольшого числа атомов — ансамбли.(1)
Появление точных методов определения поверхности катализаторов позволило установить, что активность, отнесённая к единице поверхности (удельная каталитическая активность), определяется химическим составом и очень мало зависит от структурных дислокаций. Удельная каталитическая активность различных граней кристаллов иногда различается в несколько раз. Большое влияние на активность оказывают нарушения химического состава (отклонение от стехиометрии, внедрение примесей, локальные химические образования и т.п.).
В 60-е годы промежуточное химическое взаимодействие в гетерогенном К. рассматривается преимущественно как локальное, определяемое электронной структурой отдельных атомов или ионов каталитически активного компонента на поверхности катализатора с учётом влияния ближайшего окружения. Значительную помощь в развитии этого подхода оказала обнаруженная экспериментально аналогия в действии твёрдых катализаторов, содержащих определённый металл, при гетерогенном К. и растворимых комплексов, компонентом которых является тот же металл, при гомогенном К. в растворах. При этом используются теории кристаллического поля и поля лигандов, ещё ранее успешно применявшиеся в химии комплексных соединений. Для ряда классов катализаторов и каталитических реакций установлены корреляции между каталитической активностью и энергиями связей реактантов с катализатором при промежуточном взаимодействии, облегчающие в отдельных случаях подбор катализаторов.(1)
Первые научные сведения о К. относятся к началу 19 в. В 1806 французские химики Н. Клеман и Ш. Дезорм открыли каталитическое действие окислов азота на окисление сернистого газа в камерном процессе получения серной кислоты, В 1811 русский химик К.С. Кирхгоф открыл, что разбавленные кислоты способны вызывать превращение крахмала в сахар (глюкозу); в 1814 им же было установлено, что эту реакцию может катализировать диастаза из ячменного солода, — так было положено начало изучению биологических катализаторов — ферментов. В 1818 французский химик Л. Тенар установил, что большое число твёрдых тел оказывает ускоряющее действие на разложение растворов перекиси водорода, а английский химик Г. Дэви открыл способность паров спирта и эфира окисляться кислородом на платине. В 1822 нем. химик И. Дёберейнер установил, что водород и кислород соединяются на платине при обычной температуре. За этим последовало открытие и ряда др. примеров резкого положительного действия веществ на скорость или возникновение химических реакций. Это привело к выделению особой группы явлений, названных нем. химиком Э. Мичерлихом контактными (1833) и швед. химиком И. Берцелиусом каталитическими (1835).(1)
В дальнейшем было открыто большое число каталитических реакций, и за последние 50 лет К. стал ведущим методом осуществления химических реакций в промышленности. Применение катализаторов позволяет проводить химические превращения с высокими скоростями при небольших температурах — большинство промышленных каталитических процессов без катализаторов вообще не могло бы быть реализовано. Подбирая катализаторы, можно направлять химические превращение в сторону образования определённого продукта из ряда возможных. Применение стереоспецифичных катализаторов позволяет регулировать и строение конечных продуктов, например полимеров. С помощью К. в начале 20 в. была решена проблема фиксации азота воздуха. Промотированные железные и другие катализаторы позволили преодолеть химическую инертность элементарного азота и осуществить синтез аммиака. Одновременно был разработан каталитический метод получения азотной кислоты путём окисления аммиака на платиновых сетках. На каталитических реакциях основываются современные методы получения водорода из природного газа. Каталитические методы занимают господствующее положение и в технологии нефтепереработки. Сотни миллионов тонн высококачественного моторного топлива производятся с помощью каталитических реакций крекинга, гидрокрекинга, риформинга, циклизации и изомеризации углеводородов нефти. Особенно большую роль играют каталитические методы в осуществлении процессов органического синтеза. В нашей стране впервые в мире было разработано и реализовано производство синтетического каучука, основанное на превращении этилового спирта в дивинил с помощью многокомпонентного окисного катализатора Лебедева. Каталитические методы используются для получения подавляющего большинства продуктов нефтехимического синтеза: растворителей, ароматических углеводородов, мономеров для производства синтетических каучуков, синтетических волокон и др. полимерных материалов. Катализаторы широко используются и для полимеризации.(1)
К. играет ведущую роль в химических превращениях в живой природе. Вся сложная система управления жизненными процессами в организмах основана на каталитических реакциях. Биологические катализаторы, называемые ферментами или энзимами, представляют собой вещества белковой природы с химически активными группами, часто включающими в свой состав атомы переходных элементов. По некоторым свойствам ферменты превосходят промышленные катализаторы. В СССР и за рубежом широко ведутся исследования новых типов сложных синтетических катализаторов — комплексных соединений, органических полупроводников, полимеров, характеризующихся более простым составом по сравнению с ферментами, но моделирующих в известной степени их действие. Науке о К. принадлежит существенная роль как в прогрессе химической промышленности, так и в раскрытии важнейших биологических закономерностей.(1)
МЕТОДЫ ОПРЕДЕЛЕНИЯ АКТИВНОСТИ КАТАЛИЗАТОРОВ
Наиболее существенной характеристикой любого катализатора является его активность в определенном каталитическом процессе. Выбор методики исследования активности связан с особенностями реакций, условиями эксперимента и т. д.
Мерой каталитической активности может служить скорость протекания реакции в исследуемом направлении в присутствии катализатора. Активность можно выражать так же снижением энергии активации при участии катализатора или отношением константы скорости данной реакции в присутствии катализатора к константе скорости этой же реакции без него.
При оценке активности контактных масс в производственных условиях обычно вычисляют скорость реакции по отношению к единице объема контакта по уравнениям
![]() (1)
(1)
где Gn— количество полученного целевого продукта; G — количество основного вещества, превратившегося за время т в объеме катализатора v; k — константа скорости каталитического процесса; С — движущая сила процесса, выражаемая произведением действующих концентраций исходных веществ и учитывающая тормозящее влияние продуктов реакции.(1)
Скорость реакции выражают и нарастанием концентрации продукта Сп во времени или степенью превращения в целевой продукт основного исходного вещества х (выход продукта), т.е. dCn/ dф или dx / dф.
Если скорость реакции выражать через концентрацию основного исходного вещества (реагента) С или через его начальную концентрацию Сн и общую степень превращения, то искомая величина составит – dCн / dф или – Cнdx / dф. Определяющим во всех этих случаях будет произведение к∆С (в литературе иногда вместо ∆С пишут f (C)).
При оценке самого активного компонента контактной массы следует определить активность единицы поверхности (удельная каталитическая активность). С этой целью необходимо замерить всю внутреннюю поверхность и полностью ее использовать в реакции, т.е. вести процесс в кинетической области. В этом случае скорость, реакции выражается формулами
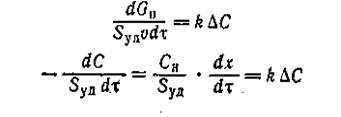 (2)
(2)
где Syд — поверхность катализатора, м2/м3 контактной массы.(1)
Существует много различных методов определения кинетических характеристик, которые могут быть разделены на две основные группы: 1) статические, осуществляемые в закрытых системах и 2) проточные — в открытых системах.
Статический метод
Реакцию проводят в замкнутом объеме до установления термодинамического равновесия, либо до полного превращения одного из исходных реагентов. Концентрация реагентов меняется от исходной до равновесной, соответственно меняется и скорость реакции по закону действующих масс (основному закону кинетики). В исследуемом объеме при отсутствии диффузионных торможений и постоянстве температуры имеют место соотношения (1)
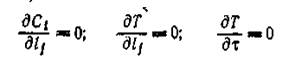 (3)
(3)
где Ci — концентрация компонента реакционной смеси; Т —температура; lj — пространственные координаты реакционной системы; — время.
Часто статический метод используют для измерения скоростей реакций, приводящих к изменению числа молекул, что позволяет следить за ходом реакции по изменению давления. На рис. 1 дана установка для изучения скорости реакции водорода с кислородом статическим методом. Перед опытом в ней устанавливают вакуум 10-5 мм рт. ст. В процессе реакции через определенные промежутки времени измеряют давление, по изменению которого рассчитывают скорость реакции.(1)
Основным преимуществом статического метода является возможность работы с очень малым количеством исходных веществ и с катализатором в любой форме, а также получение всей кинетической кривой в одном опыте, высокая чувствительность и точность измерений. Однако правильность выводов из результатов, полученных этим методом, зависит, от справедливости допущения квазистационарного протекания реакции. Статический метод рекомендуется применять в тех случаях, когда изменение состава реакционной смеси заметно не сказывается на составе и активности поверхности катализатора и когда изменение состава поверхности катализатора происходит гораздо быстрее, чем реакция.(1)
Вариантом статического метода является проведение реакций в жидкой фазе (например, гидрирование органических веществ). Из-за невысокой чувствительности метода (в отличие от статического метода в газовой фазе) обычно используют катализатор в виде зерен, порошков, но не в виде пленок или нитей. Чувствительность этого варианта статического метода значительно ниже, чем при проведении реакций в газовой фазе.(1)
К недостаткам статического метода следует отнести его интегральный характер (т. е.. необходимость проводить дифференцирование опытных данных),' возможные перепады температур и концентраций и ряд других. Поэтому указанный метод в настоящее время находит весьма ограниченное применение при изучении активности промышленных катализаторов.(1)
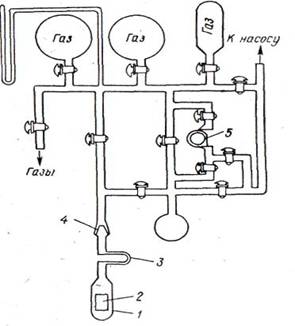
Рис. 1. Установка для изучения скорости взаимодействия водорода с кислородом: 1—контактный кварцевый аппарат; 2 —палладиевая пластинка; 3 — U-образная трубка для вымораживания ртути и паров смазки; 4—шлиф; 5—капилляр.
Проточные (динамические) методы
При исследовании катализаторов наиболее распространены проточные методы измерения каталитической активности. В проточных установках поток реагентов пропускают с определенной скоростью через реакционный объем, содержащий катализатор, производя замеры параметров процесса и анализы состава на входе в реактор, на выходе из него и, по возможности, в различных точках этого объема. Проточные методы позволяют проводить кинетические исследования в установившихся условиях, т.е. при постоянстве исходных концентраций, температур, давлений, степени перемешивания и других параметров в каждом отдельном опыте. При переходе от одного опыта к другому изменяют определенные параметры процесса на заданную величину.(1)
Наиболее распространены два типа проточного метода: проточный и проточно-циркуляционный.
Проточный метод является интегральным и непрерывным и позволяет осуществлять процесс как угодно долго при заданных концентрациях, температурах, давлениях, линейных и объемных скоростях газового потока на входе в реактор. Естественно, что концентрации реагирующих веществ и другие параметры изменяются по длине (высоте) реактора в результате химического превращения. Аппаратурное оформление таких установок проще, а чувствительность ниже, чем статических.(1)
При использовании проточного метода с неподвижным слоем катализатора в реакторе обычно допускают, что движение газа в слое катализатора отвечает режиму идеального вытеснения, т.е. пренебрегают радиальными градиентами давления, температуры, концентрации. Соответственно среднюю скорость процесса по высоте слоя Н или по времени контакта т (поскольку т пропорционально Н) определяют интегрированием кинетических уравнений (1) и (2). Аналитическое решение кинетических уравнений, как правило, возможно лишь с применением вычислительных машин. При их отсутствия прибегают к графическому дифференцированию зависимости x = f(x), что вносит погрешности.
Основным достоинством проточного метода является возможность определения каталитической активности при стационарном состоянии катализатора. Существенным недостатком — невозможность прямого измерения скорости реакции и трудность осуществления в реальных условиях режима идеального вытеснения (1).
Однако ряд преимуществ проточного метода (простота конструктивного оформления, непрерывность работы, возможность проверки катализатора в условиях, близких к производственным) обеспечили ему широкое применение при изучении каталитических реакций окисления окиси углерода, сернистого ангидрида, аммиака, спиртов и многих других. На рис. 2 дана общая схема проточной установки для определения активности катализатора в процессе окисления сернистого ангидрида (1).
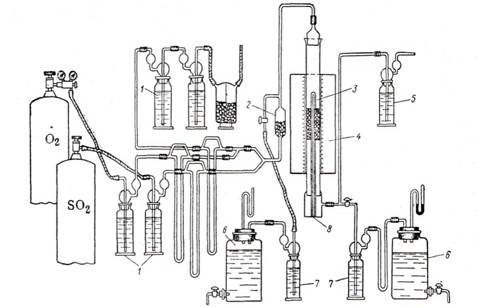
Рис. 2. Стандартная установка для испытания активности контактных масс окисления S02 проточным методом: 1—дрексель; 2—смеситель газов; 3 —контактная трубка; 4— электрическая печь; 5—поглотительная склянка с серной кислотой: 6 —аспиратор; 7—анализатор; 8 — термопара.
Газовую смесь через смеситель 2 направляют в реактор 3 с контактной массой. Контактная трубка помещена в электрическую печь 4, снабженную тремя самостоятельно регулируемыми нихромовыми спиралями. Это дает возможность регулировать температуру отдельно в разных частях слоя контактной массы с достаточным приближением к изотермичности. Колебания температуры по слою не должны превышать 5°С. Концентрацию сернистого ангидрида определяют до контактной трубки и после нее.
Скорость процесса окисления S02 в SO3 на ванадиевом катализаторе (в неподвижном слое) выражается уравнением (1)
![]() (4)
(4)
где х — степень превращения, доли ед.; т —время контакта, с; k — константа скорости реакции, с-1 • см2/кгс;. а, b — начальные концентрации сернистого ангидрида и кислорода, соответственно, объемн. %; xp — равновесная степень превращения доли ед.; Т— температура, К.
Проточно-циркуляционный метод измерения активности осуществляют путем определения концентраций компонентов в циркулирующей газовой смеси при малых степенях превращения за один проход через катализатор.
Описанные выше методы являются интегральными и их применение основано на принятии упрощающих предположений o peжиме идеального вытеснения и о квазистационарном состоянии системы. Отклонения от таких режимов обусловлены наличием определенных градиентов, возникающих в применяемых системах (1).
Безградиентный проточно-циркуляционный метод осуществляют в условиях практического отсутствия в реакционной зоне перепадов концентраций, температур, скоростей. Принцип его применительно к изучению кинетики гетерогенных каталитических реакций был впервые предложен М.И. Темкиным, С.Л. Киперманом и Л.И. Лукьяновой. Перемешивание в проточно-циркуляционной системе достигается применением интенсивной циркуляции реакционной смеси через катализатор в замкнутом объеме при непрерывном поступлении и выведении газового потока, причем количество циркулирующего газа должно значительно превышать количество вновь вводимого исходного газа. Циркуляция с большой скоростью происходит с помощью насосов: механических, поршневых или электромагнитных, мембранных и других. Циркуляционный контур, состоящий из, электромагнитного насоса (производительность 600—1000 л/ч), клапанной коробки двойного действия 2 и реактора 1 представлен на рис. 3. Высокая линейная скорость реакционной смеси в цикле и малая степень превращения обусловливают минимальные градиенты концентраций и температур, при этом слой можно рассматривать, как бесконечно малый, а реактор — как аппарат идеального смешения. Следовательно, скорость
процесса можно в данном случае определить отношениями (1)
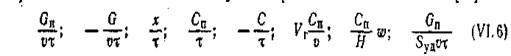 (5)
(5)
где Gn и G —количества полученного за время т продукта или превращенного исходного вещества, соответственно; Сп и С —концентрации продукта и. основного исходного вещества к моменту времени х, соответственно; w—линейная скорость газа; v — объем катализатора; Vг — объем газа; H —высота слоя катализатора.(1)