Принципы биохимических исследований
Содержание
Лекция 1. Оборудование биохимической лаборатории. Общие принципы биохимического исследования
Лекция 2. Разрушение клеток и экстракция. Центрифугирование
Лекция 3. Разделение белков путем осаждения
Лекция 4. Буферные растворы и специальные добавки. Ультрафильтрация. Диализ. Детергенты и их применение
Лекция 5. Общие принципы хроматографии, классификация хроматографических методов
Лекция 6. Материалы матриц сорбентов и обменников. Техника колоночной хроматографии
Лекция 7. Адсорбционная и распределительная хроматографии
Лекция 8. Тонкослойная хроматография
Лекция 9. Ионообменная хроматография
Лекция 10. Ионообменная ЖХВД белков. Хроматофокусирование
Лекция 11. Аффинная хроматография
Лекция 12. Гель-фильтрация
Лекция 13. Теоретические и методические основы электрофореза
Лекция 14. Изоэлектрическое фокусирование и изотахофорез
Лекция 15. Обнаружение, количественное определение и характеристика макромолекул после электрофореза
Лекция 16. Принцип иммунного электрофореза. Иммунофиксация
Лекция 17. Электросинерез. Электроиммуноанализ
Лекция 18. Методы меченых атомов
Лекция 19. Спектрофотометрические методы анализа
Лекция 20. Флюориметрические методы анализа
Лекция 21. Иммуноферментный анализ
Лекция 22. Радиометрический анализ. Масс-спектроскопия
Лекция 23. Блоттинг-анализ
Лекция 1. Оборудование биохимической лаборатории. Общие принципы биохимического исследования
Источники опасности и меры безопасности в лаборатории при проведении биохимического анализа. Особенности применения общих лабораторных методов в биохимическом эксперименте. Микро - и нанометоды.
Лабораторная посуда: материалы для её изготовления, выбор оптимального материала в зависимости от поставленной задачи биохимического эксперимента, виды лабораторной посуды, биосовместимые способы мытья и сушки лабораторной посуды, особые способы подготовки лабораторной посуды для биохимического анализа.
Исходные реактивы для биохимической лаборатории. Сведения о реактивах: маркировка реактивов, использование литературных и электронных источников справочной информации. Особенности хранения реактивов для биохимического анализа. Способы проверки качества и чистоты реактивов, выбор способа проверки, адекватного поставленной аналитической задаче. Методы дополнительной подготовки и очистки реактивов для биохимического анализа. Перекристаллизация.
Методы отбора реактивов в биохимическом анализе. Взвешивание: виды весов для аналитической биохимии, принципы и источники погрешностей взвешивания. Дозирование жидкостей, использование пипеточных дозаторов, возможные источники погрешностей. Особенности приготовления растворов в аналитической биохимии: принципы приготовления, способы выражения, концентраций, растворимости, растворители для биохимического анализа, способы постепенного добавления реактивов, растворение плохо растворимых веществ (суспендирование, эмульгирование, детергенты, использование которых допустимо в биохимическом анализе). Буферные растворы для использования в биохимическом анализе.
Методы контроля температуры в биохимической лабораторной практике.
Необходимость проведения ряда биохимических анализов в специальных условиях. Техника работ с реагентами, чувствительными к влаге, кислороды воздуха и свету. Проведение реакций в апротонных растворителях, в безводных условиях и в инертной атмосфере. Техника проведения фотохимических реакций.
Лекция 2. Разрушение клеток и экстракция. Центрифугирование
Принцип метода, основные определения и формулы. Центрифугирование применяется для разделения неоднородных жидких сред.
Центрифугирование позволяет разделить смесь, состоящую из двух или более компонентов с разной удельной плотностью, если по крайней мере один из этих компонентов - жидкость.
Разделение веществ с помощью центрифугирования основано на разном поведении частиц в центробежном поле. В центробежном поле частицы, имеющие разную плотность, форму или размеры, осаждаются с разной скоростью.
Скорость осаждения, или седиментации, зависит от центробежного ускорения (G), прямо пропорционального угловой скорости ротора (, в рад/с) и расстоянию между частицей и осью вращения (г, в см): G = 2 • г. Поскольку один оборот ротора составляет 2л радиан, угловую скорость ротора в оборотах в минуту (об. /мин) можно записать так: v = 2p60 (об. /мин), а центробежное ускорение тогда будет равно: G =4p2r/3600 (об. /мин) 2.
Центробежное ускорение обычно выражается в единицах g {гравитационная постоянная, равная 980 см*с-1) и называется относительным центробежным, ускорением (ОЦУ), т.е. ОЦУ=4p2r/3600*980 (об. /мин) 2 или ОЦУ = 1,11*10-5*r (об. /мин) 2 (*)
На основании уравнения (*) Доулом и Котциасом была составлена номограмма, выражающая зависимость ОЦУ от скорости вращения ротора и радиуса г - среднего радиуса вращения столбика жидкости в центрифужной пробирке (т.е. расстояния от оси вращения до середины столбика жидкости).
Номограмма для расчета центробежного ускорения

Для определения G соединяют прямой линией значения радиуса и скорости вращения ротора на крайних шкалах; точка пересечения этой прямой со средней шкалой дает искомую величину центробежного ускорения. Следует иметь в виду, что правая колонка цифр шкалы G соответствует правой колонке цифр шкалы скорости вращения ротора; левая - левой.
Скорость седиментации сферических частиц зависит не только от центробежного ускорения, но и от плотности и радиуса самих частиц и от вязкости среды суспендирования. Время осаждения сферической частицы в жидкой среде от мениска жидкости до дна центрифужной пробирки обратно пропорционально скорости седиментации и определяется следующим уравнением (закон Стокса, видоизмененный Сведбергом и Никольсом):
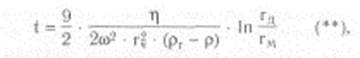
где t - время седиментации, с; h - вязкость среды, Паскаль • секунда; гч - радиус частицы, см; рч - плотность частицы (удельный вес); p - плотность среды (жидкости) или удельный вес; гм - расстояние от оси вращения до мениска жидкости, см; гд - расстояние от оси вращения до дна пробирки, см.
Как следует из уравнения (**), при заданной скорости вращения ротора время, необходимое для осаждения гомогенных сферических частиц, обратно пропорционально квадрату их радиусов и разности плотностей частиц и среды и прямо пропорционально вязкости среды. Поэтому смесь гетерогенных, приблизительно сферических частиц, различающихся по плотности и (или) размерам, можно выделить либо за счет разного времени осаждения их на дно пробирки при данном ускорении, либо за счет распределения седиментирующих частиц вдоль пробирки, устанавливающегося через определенный промежуток времени. При разделении веществ необходимо учитывать и такие важные факторы, как плотность и вязкость среды.
Описанными методами можно выделять клеточные органеллы из гомогенатов тканей. Основные компоненты клетки осаждаются в следующей последовательности: сначала целые клетки и их фрагменты, затем ядра, хлоропласты, митохондрии, лизосомы (или другие микротельца), микросомы (фрагменты гладкой и шероховатой эндоплазматической сети) и, наконец, рибосомы.
Осаждение несферических частиц не подчиняется уравнению (**), поэтому частицы одинаковой массы, но различной формы осаждаются при разных скоростях. Эта особенность используется при исследовании конформации макромолекул.
Препаративное центрифугирование заключается в выделении биологического материала для последующих биохимических исследований.
С помощью препаративного центрифугирования выделяют большое количество клеточных частиц для изучения их морфологии, структуры и биологической активности. Метод применяется для выделения таких биологических макромолекул, как ДНК и белки, из предварительно очищенных препаратов.
Аналитическое центрифугирование применяется главным образом для изучения чистых и практически чистых препаратов макромолекул или частиц, например, рибосом. В данном случае используется небольшое количество материала, а седиментация исследуемых частиц непрерывно регистрируется с помощью специальных оптических систем. Метод позволяет получать данные о чистоте, молекулярной массе и структуре материала.
В практике препаративное центрифугирование применяется гораздо чаще, чем аналитическое, поэтому мы остановимся на нем более подробно, хотя в основе обоих методов лежат общие принципы.
Лекция 3. Разделение белков путем осаждения
Осаждение нуклеиновых кислот.
Обычно, когда говорят о высаливании, имеют в виду высаливание именно сульфатом аммония. Этому методу уже более 130 лет. Раньше он применялся и для фракционирования, сейчас, в основном, как дешёвый и удобный метод осаждения белков. Можно считать, что повезло, если при этом получается ещё и существенная очистка (при высаливании из клеточного экстракта можно рассчитывать на приблизительно 2-10 кратное обогащение).
Почему именно сульфат аммония?
При равной молярной концентрации поливалентные анионы долее эффективны для высаливания, чем моновалентные (отчасти из-за того, что имеет значение не концентрация соли, а ионная сила раствора), а поливалентные катионы даже препятствуют действию поливалентных анионов. Получается, что оптимально сочетание - это поливалентный анион с моновалентными катионами.
Эффективность высаливания убывает в серии Гофмейстера (Hofmeister):
Цитрат > Сульфат > Фосфат > Хлорид > Нитрат > Тиоционат
В этом же ряду убывает стабилизирующий эффект и возрастают хаотропные свойства соли. Таким образом, наиболее подходящие кандидаты: цитрат и сульфат. Сульфат более удобен из-за лучшей растворимости (например, при нормальной температуре растворимость аммонийных солей цитрата и сульфата равны примерно 2.5М и 4.1М); низкой цены и стабилизирующего влияния, которое он оказывает на большинство белков при концентрациях выше 0.5М.
(NH4) 2SO4 преципитируют белки по двум механизмам
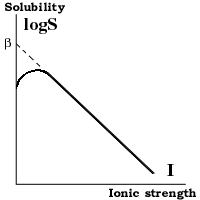
сульфат-ионы делают молекулу белка более компактной (менее растворимой) за счёт взаимодействия с положительно заряженными аминокислотами. Это взаимодействие более эффеективно при pH обезвоживание. Один ион SO42 - имеет 13-14 молекул H2O только в первом гидратном слое и, возможно, больше - во втором. Если одна молекула сульфата координирует даже 15 молекул H2O, то 3M сульфат аммония связывает 45 из имеющихся в воде 55M молекул H2O. I = 1/2 где: ci - концентрация иона; zi - заряд иона. Например, для 1M NaCl: I = 1/2 (1 (1) 2 + 1 (1) 2) = 1 для 1M (NH4) 2SO4: I = 1/2 (2 (1) 2 + 1 (1) 2) = 3 Растворимость белков Влияние ионной силы и температуры При низкой ионной силе (<0.2M) растворимость белка увеличивается при повышении концентрации соли, так как экранирование притяжения противоположно заряженных групп приводит к разрыхлению структуры белка. При высокой ионной силе (>0.2M) - растворимость белка понижается из за высаливания и обезвоживания. Она падает экспоненциально с повышением ионной силы: logS = ß - KsI, где: S (g/l) - растворимость белка; I - ионная сила; ß; Ks - константы. Ks - слегка различается для различных белков и почти не зависит от pH и температуры. ß - сильно зависит от белка, pH и температуры; повышение температуры вызывает понижение ß => уменьшение растворимости белка. Влияние pH. При низкой ионной силе (<0.2M) растворимость белка минимальна при pH равном изоэлектрической точке белка. При высоких концентрациях (NH4) 2SO4 растворимость повышается с повышением pH, так как при низких pH сульфат ион компактизует белок взаимодействуя с положительно заряженными группами. Так что лучше проводить осаждение при pH Влияние начальной концентрации белка Белки бывают двух типов. Для типа I растворимость не зависит от исходной концентрации белка; для типа II - зависит сильно. Высокая концентрация ионов аммония в осадке может мешать точному определению концентрации белка. Высаливаются не только белки, но и, например, детергенты. Например, 0.5% Tween 20 и Triton X100 начинают агрегировать при концентрациях сульфата аммония больше 1M. Образующийся преципитат имеет плотность чуть меньше плотности солевого раствора. При центрифугировании он всплывает, прихватывая с собой белки. Осаждение сульфатом амония нельзя использовать для белков, требующих присутствия Ca2+ из-за нерастворимости сульфата кальция. Буферные растворы (синоним: буферные смеси, буферные системы, буферы) - растворы с определенной концентрацией водородных ионов, содержащие сопряженную кислотно-основную пару, обеспечивающую устойчивость величины их водородного показателя при незначительных изменениях концентрации либо при добавлении небольшого количества кислоты или щелочи. Кислотно-основная пара Б. р. представляет собой слабую кислоту и ее соль, образованную сильным основанием (например, уксусная кислота СН3СООН и ацетат натрия CH3COONa) или слабое основание и его соль, образованную сильной кислотой (например, гидроокись аммония NH4OH и хлористый аммоний NH4CI). При разведении раствора или добавлении к нему некоторого количества кислоты или щелочи кислотно-основная пара способна соответственно быть донором либо акцептором водородных ионов, поддерживая Т.о. величину водородного показателя на относительно постоянном уровне. Буферные растворы сохраняют устойчивость буферных свойств в определенном интервале значений рН, то есть обладают определенной буферной емкостью. За единицу буферной емкости условно принимают емкость такого буферного раствора, для изменения рН которого на единицу требуется добавить 1 моль сильной кислоты или сильной щелочи на 1 л раствора. Буферная емкость находится в прямой зависимости от концентрации Б. р.: чем концентрированнее раствор, тем больше его буферная емкость; разведение Б. р. сильно уменьшает буферную емкость и лишь незначительно изменяет рН. Тканевая жидкость, кровь, моча и другие биологические жидкости являются буферными растворами. Благодаря действию их буферных систем поддерживается относительное постоянство водородного показателя внутренней среды, обеспечивающее полноценность метаболических процессов. Наиболее важной буферной системой является бикарбонатная система крови. Концентрация в крови бикарбонатов служит одним из основных показателей кислотно-щелочного состояния организма. Этот показатель позволяет установить характер нарушения кислотно-щелочного равновесия при ряде патологических процессов. В лабораторной практике Б. р. используют в тех случаях, когда то или иное исследование может быть проведено лишь при постоянном значении рН (например, определение активности ферментов, изучение кинетики ферментативных реакций, электрофоретическое разделение белковых смесей и др.) и в качестве стандартов при определении рН различных растворов, в т. ч. биологических жидкостей. Буферные растворы готовят обычно путем растворения в воде взятых в соответствующих пропорциях слабой кислоты и ее соли, образованной щелочным металлом, частичной нейтрализации слабой кислоты сильной щелочью или слабого основания сильной кислотой, растворения смеси солей многоосновной кислоты. Всем хроматографическим методам присущи некоторые общие характеристики, позволяющие ниже изложить элементы их обобщенной теории. Однако сначала рассмотрим специфические особенности различных вариантов хроматографического фракционирования. Это, с одной стороны, позволит за теоретическими рассуждениями все время видеть реальные черты хроматографического эксперимента, а с другой - даст возможность ввести классификацию хроматографиче-ских методов. В ходе дальнейшего изложения (в частности, для его разбиения по главам) удобнее всего классифицировать методы по основному принципу фракционирования. Такую классификацию мы рассмотрим достаточно подробно и лишь в конце раздела кратко отметим другие возможные варианты классификации. КЛАССИФИКАЦИЯ ПО ПРИНЦИПУ ФРАКЦИОНИРОВАНИЯ. Как уже упоминалось, в любом хроматографическом процессе фигурируют неподвижная и подвижная фазы, между которыми распределяются молекулы фракционируемой смеси веществ. Под основным принципом фракционирования будем подразумевать природу физического, химического или биологического явления, обусловливающего такое распределение. КЛАССИФИКАЦИЯ ПО СПОСОБУ ЭЛЮЦИИ. Фронтальный анализ. Так называется вариант хроматографического процесса, когда раствор смеси компонентов непрерывно подается на вход хроматографической колонки. На выходе ее в этом случае появляются один за другим несколько "фронтов" элюата. За первым из них следует чистый, быстрее других мигрирующий в данной системе компонент смеси, отличающийся, очевидно, наименьшим сродством к неподвижной фазе. Второй фронт отмечает добавление к нему следующего по подвижности компонента. За третьим фронтом следует уже смесь трех компонентов. В настоящее время по вполне понятным причинам фронтальный анализ почти вышел из употребления и применяется лишь в отдельных, специальных случаях. Вытеснительная хроматография. В этом варианте в колонку или на стартовую линию хроматографической пластинки наносят определенную порцию раствора исходной смеси веществ, а затем ведут элюцию раствором вещества, обладающего заведомо большим сродством к неподвижной фазе хроматографической системы, чем любой из компонентов смеси. Происходит вытеснение их из неподвижной фазы, причем в первую очередь тех, которые обладают меньшим сродством к сорбенту, а затем и всех остальных. Элюент выталкивает все компоненты смеси впереди себя наподобие поршня. Так как они выходят в подвижную фазу концентрированными, то между ними также идет конкуренция за связь с неподвижной фазой. Компоненты, уступающие другим в силе сродства к этой фазе, оттесняются еще вперед, где сорбируются, но только до тех пор, пока их опять не вытеснят компоненты, обладающие большим сродством к сорбенту. В результате такого чередования сорбции и вытеснения компоненты смеси будут выходить из колонки один за другим в порядке возрастания силы их связи с неподвижной фазой. Ясно, что при этом зоны соседних компонентов будут соприкасаться или даже немного перекрываться друг с другом. Для аналитического фракционирования метод непригоден, но хорош для препаративного или полупромышленного разделения веществ, поскольку емкость колонки здесь используется очень эффективно. Хроматографическая элюция. В отличие от предыдущего в этом методе элюирующий раствор обладает меньшим сродством к сорбенту, чем любой из компонентов вносимой на колонку или пластинку смеси веществ. Эти компоненты постепенно "вымываются" из неподвижной фазы и движутся вдоль колонки за счет непрерывного перераспределения их молекул между неподвижной фазой и элюентом. Каждый из них мигрирует независимо от других в соответствии с соотношением сил его сродства к неподвижной и подвижной фазам. Миграция идет тем медленнее, чем больше сродство к неподвижной фазе. Именно этот, пригодный для аналитических целей вариант хроматографии подробно рассмотрен в следующем разделе, поэтому здесь можно ограничиться указанием на то, что при хроматографической элюции компоненты смеси выходят из колонки отдельными, разделенными друг от друга зонами, которые в соответствии с типичной формой профиля распределения вещества в каждой такой зоне (см. ниже) часто называют хроматографическими пиками. КЛАССИФИКАЦИЯ ПО РАСПОЛОЖЕНИЮ НЕПОДВИЖНОЙ ФАЗЫ. Эта классификация не требует особых пояснений. Если пористые гранулы геля или сорбента для любого типа хроматографии заполняют стеклянную или металлическую колонку, то говорят о хроматографии на колонке, или "колоночной хроматографии", хотя последнее выражение относится к категории укоренившегося жаргона. Хроматографию на колонке во многих случаях теперь ведут при очень большом давлении подачи элюента (до 300-400 атм), что позволяет уменьшить диаметр гранул до 5-10 мкм с вытекающими отсюда (см. ниже) существенными преимуществами в быстроте и качестве фракционирования микроколичеств исходного вещества. За это приходится расплачиваться использованием дорогостоящих стальных прецизионных колонок и специальной аппаратуры, но в случае серийных анализов такие затраты себя оправдывают. Жидкостную Хроматографию на колонках при высоком давлении условимся сокращенно обозначать ЖХВД. В английской литературе принято обозначение HPLC, которое расшифровывают как "high pressure (иногда - Align performance) liquid chromatography". Если хроматографический процесс идет в наклонно расположенном, ровном и относительно толстом (несколько миллиметров), открытом с поверхности слое гранул, между которыми жидкость подвижной фазы течет только под действием силы тяжести, то его можно назвать хроматографией в толстом слое" Практически этот метод нашел себе применение только для гель-фильтрации. Тонкий (0,1-0,5 мм) слой гранул, адсорбированных или иным образом закрепленных на поверхности пластинки из стекла или пластика, позволяет осуществлять Хроматографию в тонком слое, или "тонкослойную Хроматографию" (ТСХ). Английское обозначение TLC (thin layer chromatography). Движение жидкой фазы происходит за счет капиллярных сил. Вместо тонкого слоя сорбента на основе целлюлозы можно использовать просто фильтровальную бумагу, иногда специальную - с введенными в нее ионогенными группами. Соответствующий процесс следует называть хроматографией на бумаге. Вместо бумаги для аналогичного типа хроматографии используют пленки из модифицированной целлюлозы, полиамидные пленки и т.д. - это варианты хроматографии на пленках, Пластинки, бумага или пленка могут располагаться горизонтально или вертикально; в последнем случае движение подвижной фазы может быть восходящим или нисходящим - это не играет принципиальной роли, так как оно обусловлено в основном капиллярными силами. Препараты на пластинки или бумагу чаще всего наносят в виде полоски или пятна раствора у одного края сорбента, неподалеку от уровня элюирующей жидкости, в которую этот край погружают. В последнее время для ТСХ все чаще применяют вариант кольцевой хроматографии, когда исходный препарат наносят в виде кольца, а элюция идет радиально. Матрицей называют твердую основу неподвижной хроматографической фазы. Она имеет вид сплошных или пористых гранул; последние часто представляют собой пространственную сетку линейных полимеров. Для придания материалу матрицы необходимых для хроматографии свойств его модифицируют. Модификация может представлять собой химическое присоединение ("присадку") ионогенных групп, гидрофобных молекул, биологически активных веществ или фиксацию путем адсорбции тонкого слоя растворителя. Хотя особенности хроматографического процесса определяются в основном характером модификации, физико-химические параметры матрицы могут существенно влиять на свойства неподвижной фазы. К таким параметрам относятся следующие: размеры и форма гранул и их пор; диапазон разброса этих размеров; механическая прочность материала матрицы; характер его смачивания и набухания в элюенте; химическая стойкость и инертность в условиях хроматографической элюции; реакционная способность, обеспечивающая возможность химической модификации матрицы. Одни и те же матрицы, по-разному модифицированные, могут образовывать неподвижную фазу для разных хроматографических методов, поэтому во избежание повторений в данной главе мы познакомимся с физико-химическими свойствами всех применяемых в настоящее время матриц, а их модификации (а также свойства и номенклатуру получаемых путем этих модификаций сорбентов) рассмотрим в последующих главах, посвященных различным методам хроматографии. Прежде чем перейти к анализу свойств различных матриц, укажем общие для них способы обозначения диапазона линейных размеров гранул (а для сфер - их диаметров). Этот диапазон указывают либо непосредственно в микронах, либо в виде интервала чисел "МЕШ" ("mesh"). Число МЕШ соответствует числу нитей на дюйм в сетке с квадратными ячейками, через которую просеиваются гранулы. С учетом толщины самих нитей это означает, что через сетку в 100 МЕШ пройдут все гранулы, максимальный размер которых меньше 160 мкм, через сетку в 200 МЕШ - меньше 80, 400 МЕШ - меньше 40. Таким образом, диапазон размеров гранул 40-80 мкм соответствует 200-400 МЕШ. В него попадут гранулы, которые просеиваются через сетку в 200 МЕШ, но не проходят через ячейки сетки в 400 МЕШ. Для мелких гранул, размер которых менее 40 мкм, принято обозначение - 400 МЕШ. ХРОМАТОГРАФИЧЕСКИЕ КОЛОНКИ. Колонки, изготовленные в лаборатории. В лабораторной стеклодувной мастерской несложно изготовить стеклянную колонку диаметром до 8 см и длиной до 1,5 м. Сверху колонка снабжена стандартным шлифом № 14 или. № 29, куда вставляется шлифованная пробка с капельницей. Последняя снабжена оливой малого диаметра, на которую можно надеть трубочку из силиконовой резины, куда вставляется тонкая (1-2 мм) полиэтиленовая или тефлоновая трубка от насоса. На нижний конец капельницы целесообразно надегь с некоторым перекосом кусочек полиэтиленовой трубочки так, чтобы при установке пробки на место трубочка почти касалась стенки колонки. Этим предотвращается взмучивание верхнего слоя сорбента при падении капли. Верхнюю половину поверхности шлифа смазывают силиконовой смазкой, но так, чтобы смазка не попадала внутрь колонки. В момент установки в гнездо пробку немного проворачивают. Если шлиф хорошо притерт, слой смазки должен быть прозрачным. Герметичность посадки пробки следует проверять перед каждым опытом - при неработающем насосе жидкость из колонки не должна вытекать. В более простом варианте шлифованную стеклянную пробку можно заменить на резиновую. В нижнюю часть колонки заплавляется стеклянный фильтр. Чтобы не забиваться, его поры должны быть заведомо меньшего размера, чем гранулы сорбента. Вместе с тем нежелательно, чтобы фильтр представлял собой большое сопротивление току элюента. Для сорбентов, используемых при обычной хроматографии низкого давления, с гранулами не мельче 30 мкм в поперечнике подходит стеклянный фильтр № 3. Однако он может постепенно забиваться, если используется сорбент, засоренный "пылью", образующейся при его истирании. Во избежание этого не следует пренебрегать описанной ниже операцией "отмучивания" сорбента. Следует также помнить о том, что стеклянный фильтр сорбирует некоторое количество белка или нуклеиновой кислоты. С этой точки зрения в качестве фильтров следует предпочесть полиамидные (найлон) или тефлоновые пористые пластинки. Однако колонку в этом случае придется делать сборной, что значительно усложняет ее конструкцию. Такие фильтры используются в продажных фирменных колонках. Под фильтром, при переходе к сливной трубке малого диаметра образуется коническая полость. При изготовлении колонки надо стремиться к тому, чтобы объем этой полости был минимальным, так как в ней может происходить смешивание близко идущих фракций. На сливной трубке имеется такая же, как на пробке, олива для трубочки из силиконовой резины, куда вставляется тонкая трубка, идущая к денситометру. Резиновую трубочку удобно пережимать винтовым зажимом, "запирая" таким образом выход из колонки. В адсорбционной хроматографии разделение веществ, входящих в смесь и движущихся по колонке в потоке растворителя, происходит за счёт их различной способности адсорбироваться и десорбироваться на поверхности адсорбента с развитой поверхностью, например, силикагеля. В распределительной ВЭЖХ разделение происходит за счет разной растворимости разделяемых веществ в неподвижной фазе, как правило, химически привитой к поверхности неподвижного носителя, и подвижной фазе - растворителе. Этот метод разделения наиболее популярен, особенно в случае, когда привитая фаза представляет собой неполярный алкильный остаток от C8 до C18, а подвижная фаза более полярна, например смесь метанола или ацетонитрила с водой. Это так называемая обращённо-фазная (обратно-фазная, или с обращением фаз) хроматография. Тонкослойная хроматография (ТСХ) первоначально была разработана для разделения липидов. Хотя хроматография на бумаге быстрее, чем хроматография на колонке, к недостаткам ее следует отнести то, что бумага может быть изготовлена только из материалов на основе целлюлозы, что не позволяет применять ее для разделения неполярных веществ. Тонкослойная хроматография сохраняет все преимущества хроматографии на бумаге, но при этом позволяет использовать любой материал, который можно тонко измельчить и получить затем однородный слой. Это могут быть неорганические вещества, например силикагель, окись алюминия, диатомовая земля и силикат магния, а также органические вещества, в частности целлюлоза, полиамиды и порошок полиэтилена. Пластинку с закрепленным сорбентом помещают в камеру, содержащую растворитель, и проявляют восходящей хроматографией. После того как фронт растворителя почти достигнет верхнего края, пластинку вынимают из камеры и сушат. Для получения двумерной хроматограммы высушенную пластинку можно повторно хроматографировать под прямым углом в другом растворителе. Положение пятен, как и при хроматографии на бумаге, определяют по окраске, по флуоресценции или при опрыскивании различными реагентами, которые реагируют с веществами в пятне с образованием окрашенных продуктов. Обычно используют следующие реагенты: нингидрин для аминокислот, родамин В для липидов, хлорид сурьмы для стероидов и терпенов, серную кислоту с последующим нагреванием практически для всех органических соединений (происходит обугливание), перманганат калия в серной кислоте для углеводородов, анисовый альдегид в серной кислоте для углеводов, пары брома для олефинов и т.д. Вещества можно элюировать путем соскребания. ИОНООБМЕННАЯ ХРОМАТОГРАФИЯ, жидкостная хроматография, основанная на разл. способности разделяемых ионов к ионному обмену с фиксир. ионами сорбента, образующимися в результате диссоциации ионогенных групп последнего. Для разделения катионов используют катиониты, для разделения анионов - аниониты (см. Иониты). Элюентом в первом случае служит р-р кислоты, во втором - р-р щелочи. Разделение ионов регулируют подбором оптим. значений рН элюента. Сильнокислотные сульфокатиониты и высокоосновные аниониты могут использоваться при любых значениях рН, слабокислотные карбоксильные катиониты - только при рН > 6; слабоосновные аниониты находятся в ионизованном состоянии при рН < 8. Варьируя рН элюента, можно резко изменять степень ионизации компонентов разделяемой смеси (сорбатов) и, следовательно, время их удерживания, добиваясь необходимой селективности разделения. Многозарядные ионы удерживаются ионитом сильнее однозарядных. При равных величинах зарядов удерживание падает с ростом радиуса гидратир. иона. Поэтому при И. х. в разб. р-рах на сульфокатионитах время удерживания катионов падает в ряду: Ва2+ > РЬ2+ > Sr2+ > Са2+ > Ni2+ > Cd2+ > Сu2+ > Со2+ > Zn2+ > Mg2+ > UO2+ > Тl+ > Ag+ > Cs+ > Rb+ > K+ > NH4+ > Na+ > H+ > Li+. Для орг. ионов на электростатич. взаимод. с фиксир. зарядами ионита накладывается еще и гидрофобное взаимод. орг. части иона с матрицей ионита. Чтобы уменьшить его вклад в удерживание орг. ионов и добиться оптим. селективности их разделения, к водному элюенту добавляют орг. компонент (1-25% метанола, изопропанола, ацетонитрила или диоксана). Элюент в И. х. кроме к-ты или основания и орг. добавок может содержать нейтральный электролит, напр. NaNO3, ионы к-рого конкурируют с разделяемыми ионами за взаимод. с сорбентом; при этом удерживание однозарядных ионов падает пропорционально концентрации соли в р-ре, двухзарядных ионов - пропорционально ее квадрату. Важна также природа нейтрального электролита: чем выше сродство его ионов к сорбенту, тем выше элюирующая сила р-ра. В И. х. анионов часто используют фосфатные р-ры, к-рые обладают большой элюирующей способностью при высоких значениях рН, где фосфат приобретает заряд - 3. Мн. неорг. катионы разделяют на сульфокатионитах, используя в качестве элюента комплексообразователи (орг. к-ты или гидроксикислоты). Разделение основано на том, что константы устойчивости образующихся комплексов, а значит и их сорбционные св-ва, даже таких близких по св-вам катионов, как лантаноиды и актиноиды, при определенных значениях рН различаются достаточно сильно; при этом заряд комплекса можно менять (вплоть до отрицательного). С помощью И. х. разделяют нек-рые нейтральные соед., если они способны превращ. в заряженные комплексы, как, напр., комплексы углеводов с борат-ионом. Удерживание разделяемых ионов в колонке пропорционально обменной емкости ионита. Для используемых в И. х. полимерных ионитов емкость 3-6 мг-экв/г, для ионитов на основе силикагеля с привитыми к его пов-сти функц. группами - на порядок ниже. При равном размере зерен (обычно 5-15 мкм) иониты на основе силикагеля обладают более высокой скоростью ионного обмена, что повышает эффективность хроматографич. колонок, однако их гидролитич. устойчивость при рН / 8 недостаточна. Для увеличения эффективности (числа теоретич. тарелок) колонки с полимерными ионитами обычно используют при повыш. т-рах (50-80 °С); при этом увеличиваются коэф. диффузии ионов в фазе ионита. В качестве сорбентов для И. х. могут использоваться нейтральные носители, пропитанные жидкими ионитами, т.е. несмешивающимися с водой орг. основаниями или к-тами, напр., триоктиламином, триоктилметиламмонием, алкиловыми зфирами алкилфосфорной к-ты. Разбавленные р-ры ионогенных ПАВ в сочетании с нейтральными гидрофобными носителями находят применение в ион-парной хроматографии (см. Жидкостная хроматография),к-рая отличается высокой эффективностью и большим числом варьируемых параметров для подбора оптим. селективности разделения. Детектирование в И. х. осуществляют с помощью любого детектора, применяемого в жидкостной хроматографии (см. Детекторы хроматографические). Наиб. универсален для ионных соединений кондуктометр, на применении к-рого основан вариант И. х. - ионная хроматография.И. х. применяется для разделения катионов металлов, напр., смесей лантаноидов и актиноидов, Zr и Hf Мо и W, Nb и Та; последние разделяют на анионитах в виде анионных хлоридных комплексов в р-рах соляной и плавиковой к-т. Щелочные металлы разделяют на катионитах в водных и водно-орг. средах, щел. - зем. и редкоземельные металлы на катионитах в присут. комплексонов. Большое значение имеет автоматич. анализ смесей прир. аминокислот на тонкодисперсном сульфокатионите в цитратном буфере при повыш. т-ре. Аминокислоты детектируют фотометрически после их р-ции с нингидрином или флюориметрически после дериватизации фталевым альдегидом. Высокоэффективная И. х. (колонки, упакованные сорбентом с размером зерен 5-10 мкм, давление для прокачивания элюента до 107 Па) смесей нуклеотидов, нуклеозидов, пурияовых и пиримидиновых оснований и их метаболитов в биол. жидкостях (плазма крови, моча, лимфа и др
![]() ci (zi) 2,
ci (zi) 2, Лекция 4. Буферные растворы и специальные добавки. Ультрафильтрация. Диализ. Детергенты и их применение
Лекция 5. Общие принципы хроматографии, классификация хроматографических методов
Лекция 6. Материалы матриц сорбентов и обменников. Техника колоночной хроматографии
Лекция 7. Адсорбционная и распределительная хроматографии
Лекция 8. Тонкослойная хроматография
Лекция 9. Ионообменная хроматография